

Comprehensive characterization of high-risk coding and non-coding single nucleotide polymorphisms of human CXCR4 gene
- PMID: 39715225
- PMCID: PMC11665994
- DOI: 10.1371/journal.pone.0312733
Comprehensive characterization of high-risk coding and non-coding single nucleotide polymorphisms of human CXCR4 gene
Abstract
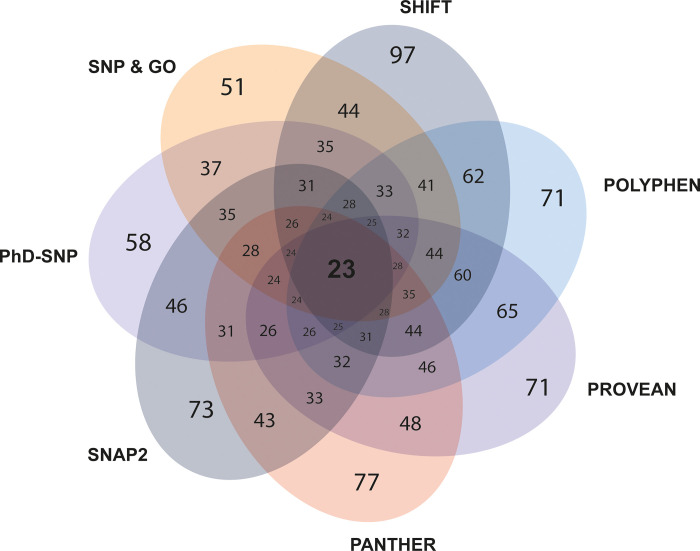
CXCR4, a chemokine receptor known as Fusin or CD184, spans the outer membrane of various human cells, including leukocytes. This receptor is essential for HIV infection as well as for many vital cellular processes and is implicated to be associated with multiple pathologies, including cancers. This study employs various computational tools to investigate the molecular effects of disease-vulnerable germ-line missense and non-coding SNPs of the CXCR4 gene. In this investigation, the tools SIFT, PROVEAN, PolyPhen-2, PANTHER, SNAP 2.0, PhD-SNP, and SNPs&GO were used to predict potentially harmful and disease-causing nsSNPs in CXCR4. Additionally, their impact on protein stability was examined by I-mutant 3.0, MUpro, Consurf, and Netsurf 2.0, combined with conservation and solvent accessibility analyses. Structural analysis with normal and mutant residues of the protein harboring these disease-associated functional SNPs was conducted using TM-align and SWIS MODEL, with visualization aided by PyMOL and the BIOVINA Discovery Studio Visualizer. The functional impact of wild-type and mutated CXCR4 variants was evaluated through molecular docking with its natural ligand CXCR4-modulator 1, using the PyRx tool. Non-coding SNPs in the 3' -UTR were investigated for their regulatory effects on miRNA binding sites using PolymiRTS. Five non-coding SNPs were identified in the 3'-UTR that can disrupt existing miRNA binding sites or create new ones. Non-coding SNPs in the 5' and 3'-UTRs, as well as in intronic regions, were also examined for their potential roles in gene expression regulation. Furthermore, RegulomeDB databases were employed to assess the regulatory potential of these non-coding SNPs based on chromatin state and protein binding regulation. In the mostly annotated variant (ENSP00000241393) of the CXCR4 gene, we found 23 highly deleterious and pathogenic nsSNPs and these were selected for in-depth analysis. Among the 23 nsSNPs, five (G55V, H79P, L80P, H113P, and P299L) displayed notable structural alternation, with elevated RMSD values and reduced TM (TM-score) values. A molecular docking study revealed the significant impact of the H113P variant on the protein-ligand binding affinity, supported by MD simulation over 100 nanoseconds, which highlighted substantial stability differences between wild-type and H113P mutated proteins during ligand binding. This comprehensive analysis shed light on the potential functional consequences of genetic variation in the CXCR genes, offering valuable insights into the implications of disease susceptibility and may pave the way for future therapeutic interventions.
Copyright: © 2024 Sarkar et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures








Similar articles
-
A computational in silico approach to predict high-risk coding and non-coding SNPs of human PLCG1 gene.PLoS One. 2021 Nov 18;16(11):e0260054. doi: 10.1371/journal.pone.0260054. eCollection 2021. PLoS One. 2021. PMID: 34793541 Free PMC article.
-
Investigating the functional and structural effect of non-synonymous single nucleotide polymorphisms in the cytotoxic T-lymphocyte antigen-4 gene: An in-silico study.PLoS One. 2025 Jan 24;20(1):e0316465. doi: 10.1371/journal.pone.0316465. eCollection 2025. PLoS One. 2025. PMID: 39854591 Free PMC article.
-
Comprehensive Characterization of the Coding and Non-Coding Single Nucleotide Polymorphisms in the Tumor Protein p63 (TP63) Gene Using In Silico Tools.Biomolecules. 2021 Nov 20;11(11):1733. doi: 10.3390/biom11111733. Biomolecules. 2021. PMID: 34827731 Free PMC article.
-
Determination of deleterious single-nucleotide polymorphisms of human LYZ C gene: an in silico study.J Genet Eng Biotechnol. 2022 Jul 1;20(1):92. doi: 10.1186/s43141-022-00383-8. J Genet Eng Biotechnol. 2022. PMID: 35776277 Free PMC article.
-
Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: current and developing technologies.Mutat Res. 2008 Jul-Aug;659(1-2):147-57. doi: 10.1016/j.mrrev.2008.05.001. Epub 2008 May 4. Mutat Res. 2008. PMID: 18565787 Free PMC article. Review.
References
-
- Collins FS, Patrinos A, Jordan E, Chakravarti A, Gesteland R, Walters LR. New goals for the U.S. Human Genome Project: 1998–2003. Science (80-). 1998;282(5389):682–9. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
