

Inter-chromosomal transcription hubs shape the 3D genome architecture of African trypanosomes
- PMID: 39715762
- PMCID: PMC11666725
- DOI: 10.1038/s41467-024-55285-9
Inter-chromosomal transcription hubs shape the 3D genome architecture of African trypanosomes
Abstract
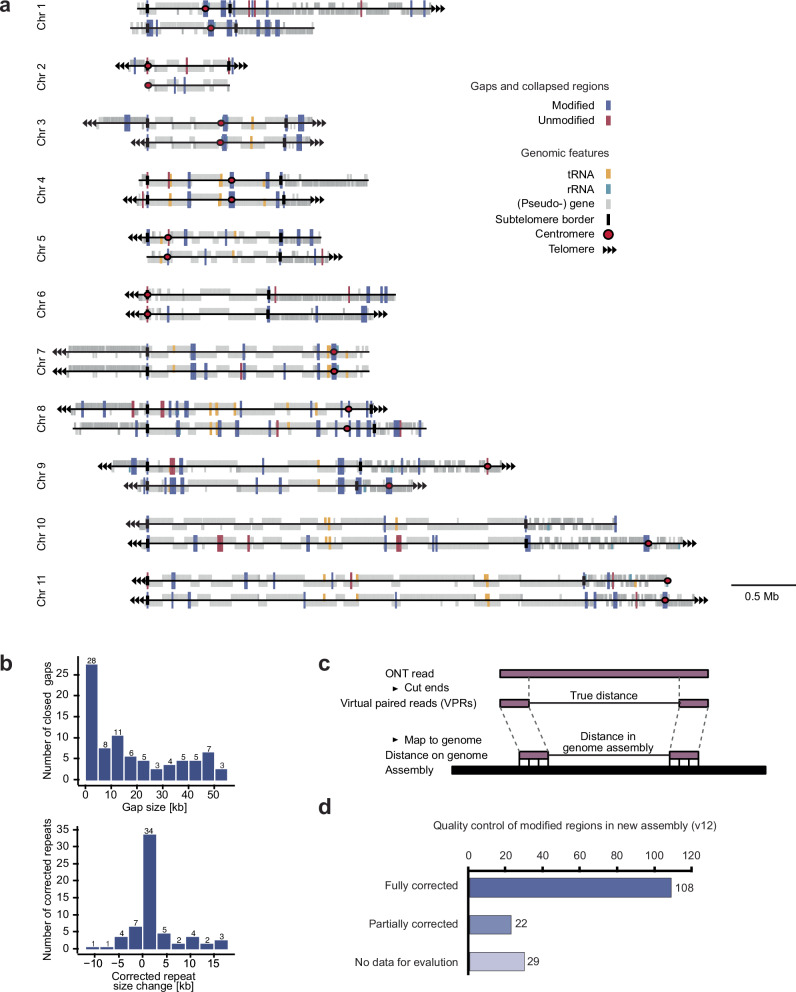
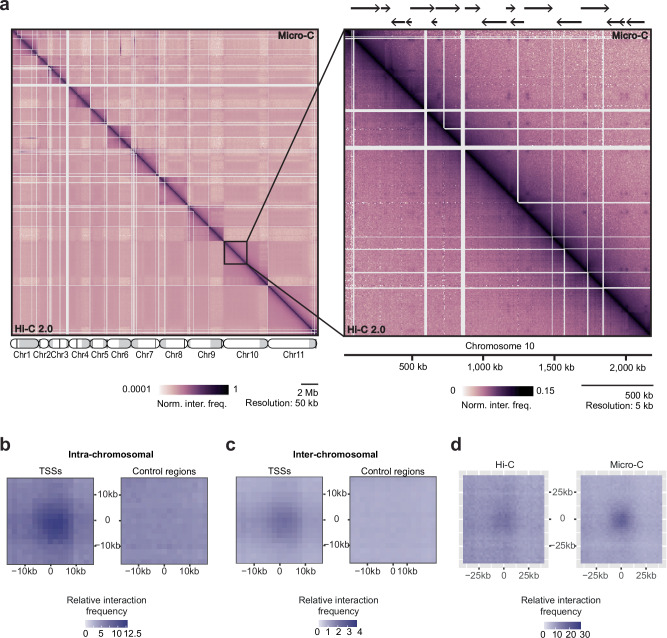
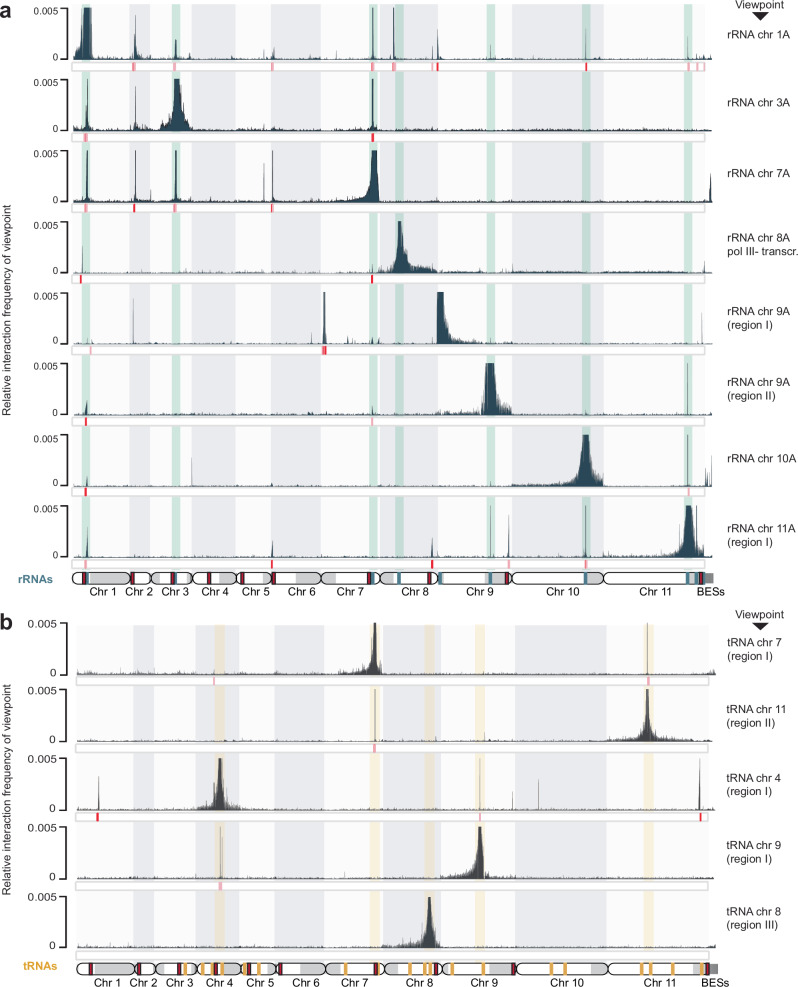
The eukaryotic nucleus exhibits a highly organized 3D genome architecture, with RNA transcription and processing confined to specific nuclear structures. While intra-chromosomal interactions, such as promoter-enhancer dynamics, are well-studied, the role of inter-chromosomal interactions remains poorly understood. Investigating these interactions in mammalian cells is challenging due to large genome sizes and the need for deep sequencing. Additionally, transcription-dependent 3D topologies in mixed cell populations further complicate analyses. To address these challenges, we used high-resolution DNA-DNA contact mapping (Micro-C) in Trypanosoma brucei, a parasite with continuous RNA polymerase II (RNAPII) transcription and polycistronic transcription units (PTUs). With approximately 300 transcription start sites (TSSs), this genome organization simplifies data interpretation. To minimize scaffolding artifacts, we also generated a highly contiguous phased genome assembly using ultra-long sequencing reads. Our Micro-C analysis revealed an intricate 3D genome organization. While the T. brucei genome displays features resembling chromosome territories, its chromosomes are arranged around polymerase-specific transcription hubs. RNAPI-transcribed genes cluster, as expected from their localization to the nucleolus. However, we also found that RNAPII TSSs form distinct inter-chromosomal transcription hubs with other RNAPII TSSs. These findings highlight the evolutionary significance of inter-chromosomal transcription hubs and provide new insights into genome organization in T. brucei.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
- SI 1610 / 2-2 and 213249687/Deutsche Forschungsgemeinschaft (German Research Foundation)
- MR/T04702X/1/RCUK | Medical Research Council (MRC)
- 221717/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- 715466 and 101044320/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
LinkOut - more resources
Full Text Sources
