

Rnalib: a Python library for custom transcriptomics analyses
- PMID: 39718766
- PMCID: PMC11734754
- DOI: 10.1093/bioinformatics/btae751
Rnalib: a Python library for custom transcriptomics analyses
Abstract
Motivation: The efficient and reproducible analysis of high-throughput sequencing datasets necessitates the development of methodical and robust computational pipelines that integrate established and bespoke bioinformatics analysis tools, often written in high-level programming languages such as Python. Despite the increasing availability of programming libraries for genomics, there is a noticeable lack of tools specifically focused on transcriptomics. Key tasks in this area include the association of gene features (e.g. transcript isoforms, introns or untranslated regions) with relevant subsections of (large) genomics datasets across diverse data formats, as well as efficient querying of these data based on genomic locations and annotation attributes.
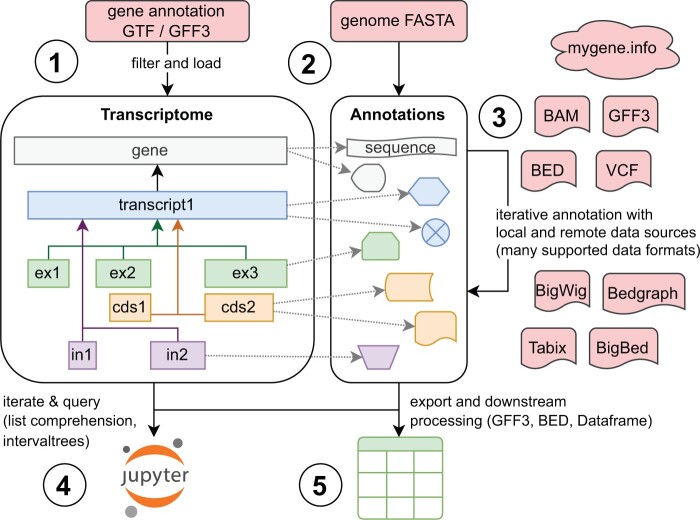
Results: To address the needs of transcriptomics data analyses, we developed rnalib, a Python library designed for creating custom bioinformatics analysis methods. Built on existing Python libraries like pysam and pyBigWig, rnalib offers random access support, enabling efficient access to relevant subregions of large, genome-wide datasets. Rnalib extends the filtering and access capabilities of these libraries and includes additional checks to prevent common errors when integrating genomics datasets. The library is centred on an object-oriented Transcriptome class that provides methods for stepwise annotation of gene features with both, local and remote data sources. The rnalib Application Programming Interface cleanly separates immutable genomic locations from associated, mutable data, and offers a wide range of methods for iterating, querying, and exporting collated datasets. Rnalib establishes a fast, readable, reproducible, and robust framework for developing novel transcriptomics data analysis tools and methods.
Availability and implementation: Source code, documentation, and tutorials are available at https://github.com/popitsch/rnalib.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
Using R and Bioconductor in Clinical Genomics and Transcriptomics.J Mol Diagn. 2020 Jan;22(1):3-20. doi: 10.1016/j.jmoldx.2019.08.006. Epub 2019 Oct 9. J Mol Diagn. 2020. PMID: 31605800 Review.
-
htsint: a Python library for sequencing pipelines that combines data through gene set generation.BMC Bioinformatics. 2015 Sep 24;16:307. doi: 10.1186/s12859-015-0729-3. BMC Bioinformatics. 2015. PMID: 26399714 Free PMC article.
-
DNA Features Viewer: a sequence annotation formatting and plotting library for Python.Bioinformatics. 2020 Aug 1;36(15):4350-4352. doi: 10.1093/bioinformatics/btaa213. Bioinformatics. 2020. PMID: 32637988
-
Bigtools: a high-performance BigWig and BigBed library in Rust.Bioinformatics. 2024 Jun 3;40(6):btae350. doi: 10.1093/bioinformatics/btae350. Bioinformatics. 2024. PMID: 38837370 Free PMC article.
-
Scalable transcriptomics analysis with Dask: applications in data science and machine learning.BMC Bioinformatics. 2022 Nov 30;23(1):514. doi: 10.1186/s12859-022-05065-3. BMC Bioinformatics. 2022. PMID: 36451115 Free PMC article. Review.
References
-
- Heger A, Marshall J, Jacobs K. et al. Pysam: Htslib interface for python. 2009. https://github.com/pysam-developers/pysam (12 December 2024, date last accessed).
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
