

Proteomics and machine learning: Leveraging domain knowledge for feature selection in a skeletal muscle tissue meta-analysis
- PMID: 39720035
- PMCID: PMC11667615
- DOI: 10.1016/j.heliyon.2024.e40772
Proteomics and machine learning: Leveraging domain knowledge for feature selection in a skeletal muscle tissue meta-analysis
Abstract
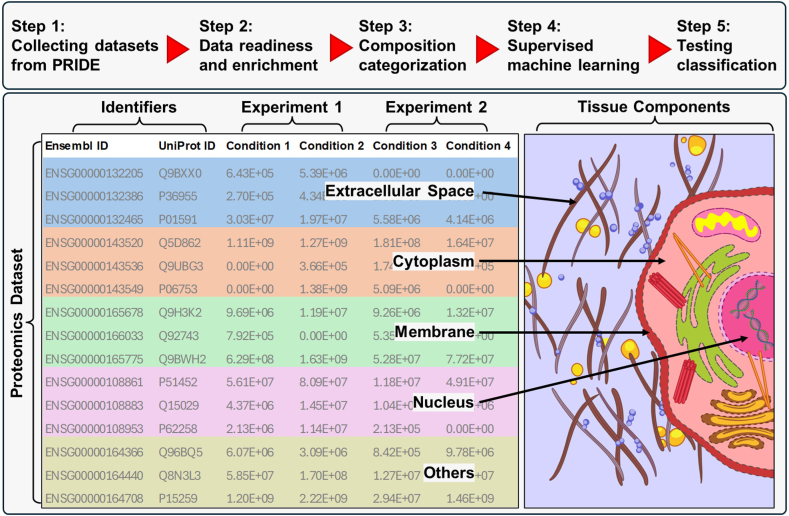
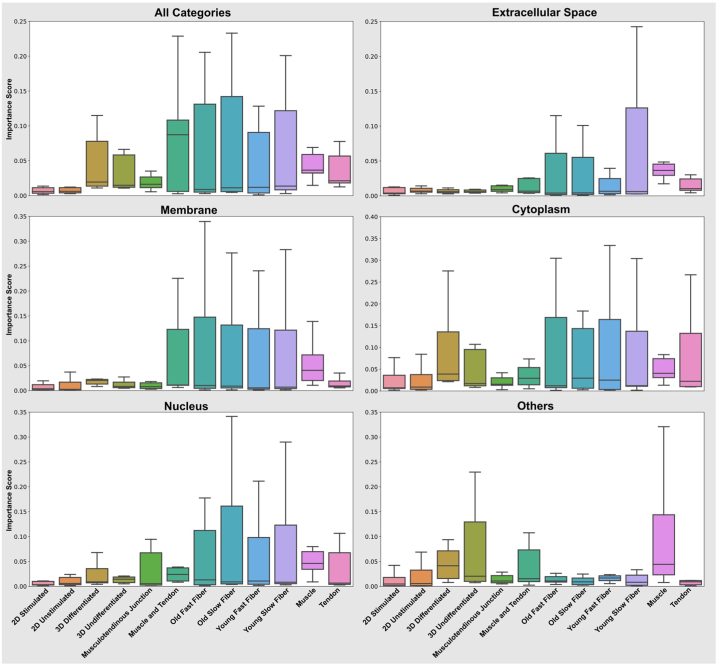
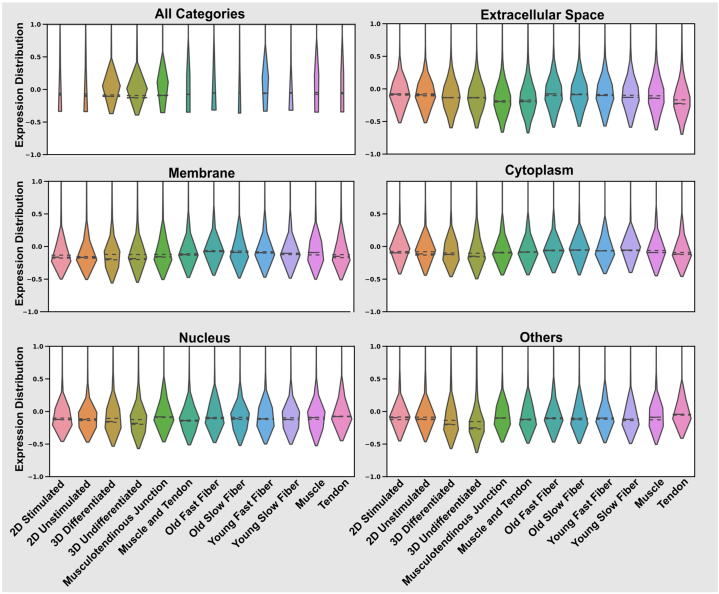
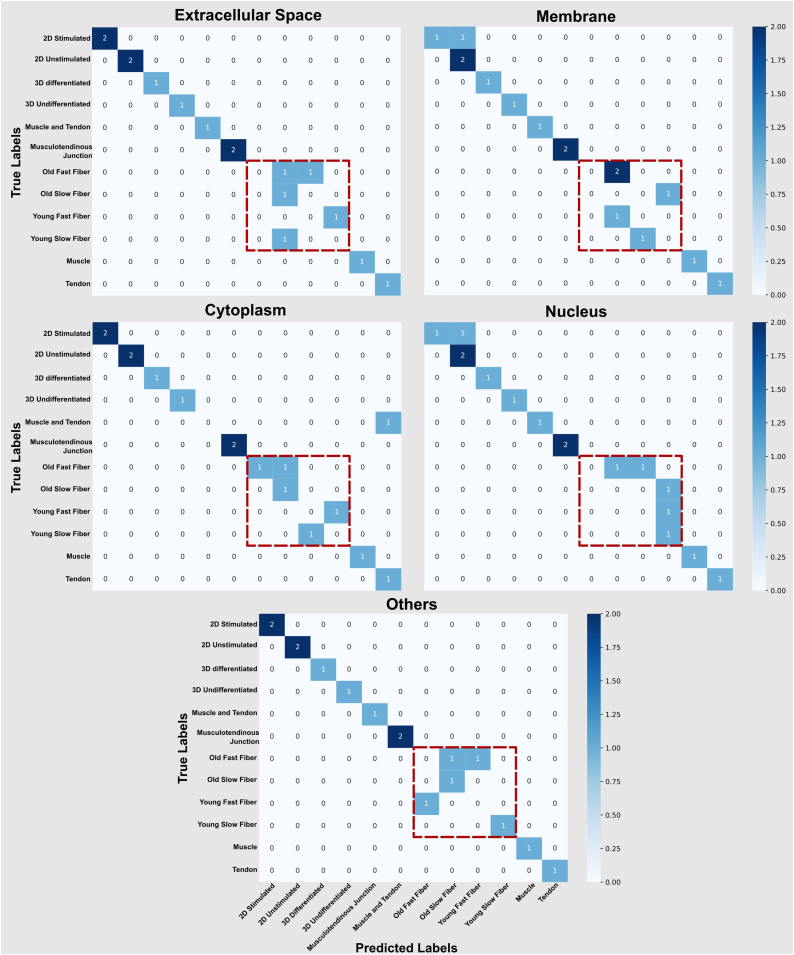
Omics techniques, such as proteomics, contain crucial data for understanding biological processes, but they remain underutilized due to their high dimensionality. Typically, proteomics research focuses narrowly on using a limited number of datasets, hindering cross-study comparisons, a problem that can potentially be addressed by machine learning. Despite this potential, machine learning has seen limited adoption in the field of proteomics. Here, skeletal muscle proteomics datasets from five separate studies were combined. These studies included conditions such as in vitro models (both 2D and 3D), in vivo skeletal muscle tissue, and adjacent tissues such as tendons. The collected data was preprocessed using MaxQuant, and then enriched using a Python script fetching structural and compositional details from UniProt and Ensembl databases. This was used to handle high-dimensional and sparsely labeled dataset by breaking it down into five smaller categories using cellular composition information and then training a Random Forest model for each category separately. Using biological context for interpreting the data resulted in improved model performance and made tailored analysis possible by reducing the dimensionality and increasing signal-to-noise ratio as well as only preserving biologically relevant features in each category. This integration of domain knowledge into data analysis and model training facilitated the discovery of new patterns while ensuring the retention of critical details, often overlooked when blind feature selection methods are used to exclude proteins with minimal expressions or variances. This approach was shown to be suitable for performing diverse analyses on individual as well as combined datasets within a broader biological context, ultimately leading to the identification of biologically relevant patterns. Besides from generating new biological insights, this approach can be used to perform tasks such as biomarker discovery, cluster analysis, classification, and anomaly detection more accurately, but incorporation of more datasets is needed to further expand the computational capabilities of such models in clinical settings.
Keywords: Domain knowledge; Feature selection; Machine learning; Proteomics; Skeletal muscle tissue.
© 2024 The Authors.
Conflict of interest statement
The authors declare no conflict of interest. All expenses are covered by the authors’ institution, Evolved.Bio.
Figures






References
LinkOut - more resources
Full Text Sources
