

Deciphering regulatory architectures of bacterial promoters from synthetic expression patterns
- PMID: 39724021
- PMCID: PMC11709304
- DOI: 10.1371/journal.pcbi.1012697
Deciphering regulatory architectures of bacterial promoters from synthetic expression patterns
Abstract
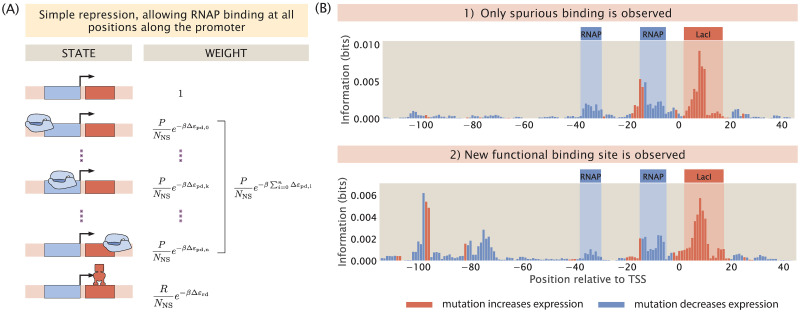
For the vast majority of genes in sequenced genomes, there is limited understanding of how they are regulated. Without such knowledge, it is not possible to perform a quantitative theory-experiment dialogue on how such genes give rise to physiological and evolutionary adaptation. One category of high-throughput experiments used to understand the sequence-phenotype relationship of the transcriptome is massively parallel reporter assays (MPRAs). However, to improve the versatility and scalability of MPRAs, we need a "theory of the experiment" to help us better understand the impact of various biological and experimental parameters on the interpretation of experimental data. These parameters include binding site copy number, where a large number of specific binding sites may titrate away transcription factors, as well as the presence of overlapping binding sites, which may affect analysis of the degree of mutual dependence between mutations in the regulatory region and expression levels. To that end, in this paper we create tens of thousands of synthetic gene expression outputs for bacterial promoters using both equilibrium and out-of-equilibrium models. These models make it possible to imitate the summary statistics (information footprints and expression shift matrices) used to characterize the output of MPRAs and thus to infer the underlying regulatory architecture. Specifically, we use a more refined implementation of the so-called thermodynamic models in which the binding energies of each sequence variant are derived from energy matrices. Our simulations reveal important effects of the parameters on MPRA data and we demonstrate our ability to optimize MPRA experimental designs with the goal of generating thermodynamic models of the transcriptome with base-pair specificity. Further, this approach makes it possible to carefully examine the mapping between mutations in binding sites and their corresponding expression profiles, a tool useful not only for developing a theory of transcription, but also for exploring regulatory evolution.
Copyright: © 2024 Pan et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures














Update of
-
Deciphering regulatory architectures from synthetic single-cell expression patterns.ArXiv [Preprint]. 2024 Jun 5:arXiv:2401.15880v2. ArXiv. 2024. Update in: PLoS Comput Biol. 2024 Dec 26;20(12):e1012697. doi: 10.1371/journal.pcbi.1012697. PMID: 38351929 Free PMC article. Updated. Preprint.
-
Deciphering regulatory architectures from synthetic single-cell expression patterns.bioRxiv [Preprint]. 2024 Jun 5:2024.01.28.577658. doi: 10.1101/2024.01.28.577658. bioRxiv. 2024. Update in: PLoS Comput Biol. 2024 Dec 26;20(12):e1012697. doi: 10.1371/journal.pcbi.1012697. PMID: 38352569 Free PMC article. Updated. Preprint.
Similar articles
-
Deciphering regulatory architectures from synthetic single-cell expression patterns.bioRxiv [Preprint]. 2024 Jun 5:2024.01.28.577658. doi: 10.1101/2024.01.28.577658. bioRxiv. 2024. Update in: PLoS Comput Biol. 2024 Dec 26;20(12):e1012697. doi: 10.1371/journal.pcbi.1012697. PMID: 38352569 Free PMC article. Updated. Preprint.
-
Deciphering regulatory architectures from synthetic single-cell expression patterns.ArXiv [Preprint]. 2024 Jun 5:arXiv:2401.15880v2. ArXiv. 2024. Update in: PLoS Comput Biol. 2024 Dec 26;20(12):e1012697. doi: 10.1371/journal.pcbi.1012697. PMID: 38351929 Free PMC article. Updated. Preprint.
-
Combining TSS-MPRA and sensitive TSS profile dissimilarity scoring to study the sequence determinants of transcription initiation.Nucleic Acids Res. 2023 Aug 25;51(15):e80. doi: 10.1093/nar/gkad562. Nucleic Acids Res. 2023. PMID: 37403796 Free PMC article.
-
Decoding enhancers using massively parallel reporter assays.Genomics. 2015 Sep;106(3):159-164. doi: 10.1016/j.ygeno.2015.06.005. Epub 2015 Jun 10. Genomics. 2015. PMID: 26072433 Free PMC article. Review.
-
Analysis of mechanisms of activation and repression at bacterial promoters.Methods. 2009 Jan;47(1):6-12. doi: 10.1016/j.ymeth.2008.10.012. Epub 2008 Oct 24. Methods. 2009. PMID: 18952178 Review.
Cited by
-
The Environment-Dependent Regulatory Landscape of the E. coli Genome.bioRxiv [Preprint]. 2025 May 15:2025.05.13.653802. doi: 10.1101/2025.05.13.653802. bioRxiv. 2025. PMID: 40462920 Free PMC article. Preprint.
-
The Environment-Dependent Regulatory Landscape of the E. coli Genome.ArXiv [Preprint]. 2025 May 13:arXiv:2505.08764v1. ArXiv. 2025. PMID: 40463697 Free PMC article. Preprint.
-
Applications of high-throughput reporter assays to gene regulation studies.Curr Opin Struct Biol. 2025 Jun 27;94:103105. doi: 10.1016/j.sbi.2025.103105. Online ahead of print. Curr Opin Struct Biol. 2025. PMID: 40580800 Review.
-
A generalized theoretical framework to investigate multicomponent actin dynamics.bioRxiv [Preprint]. 2024 Dec 12:2024.12.10.627743. doi: 10.1101/2024.12.10.627743. bioRxiv. 2024. PMID: 39713386 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
