

Computational Analysis of MDR1 Variants Predicts Effect on Cancer Cells via their Effect on mRNA Folding
- PMID: 39724131
- PMCID: PMC11670953
- DOI: 10.1371/journal.pcbi.1012685
Computational Analysis of MDR1 Variants Predicts Effect on Cancer Cells via their Effect on mRNA Folding
Abstract
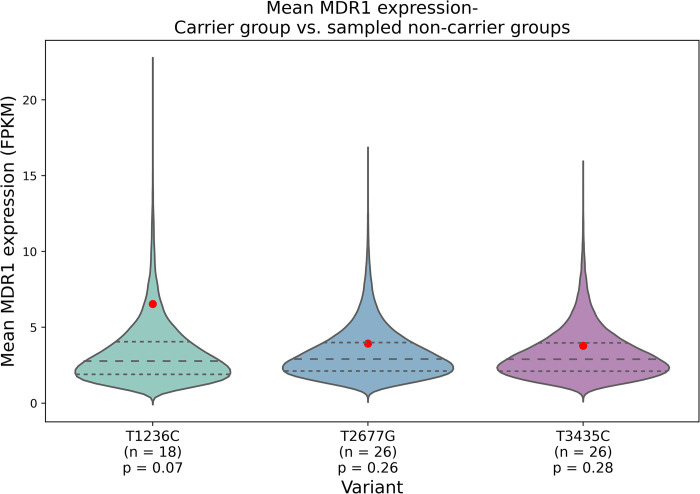
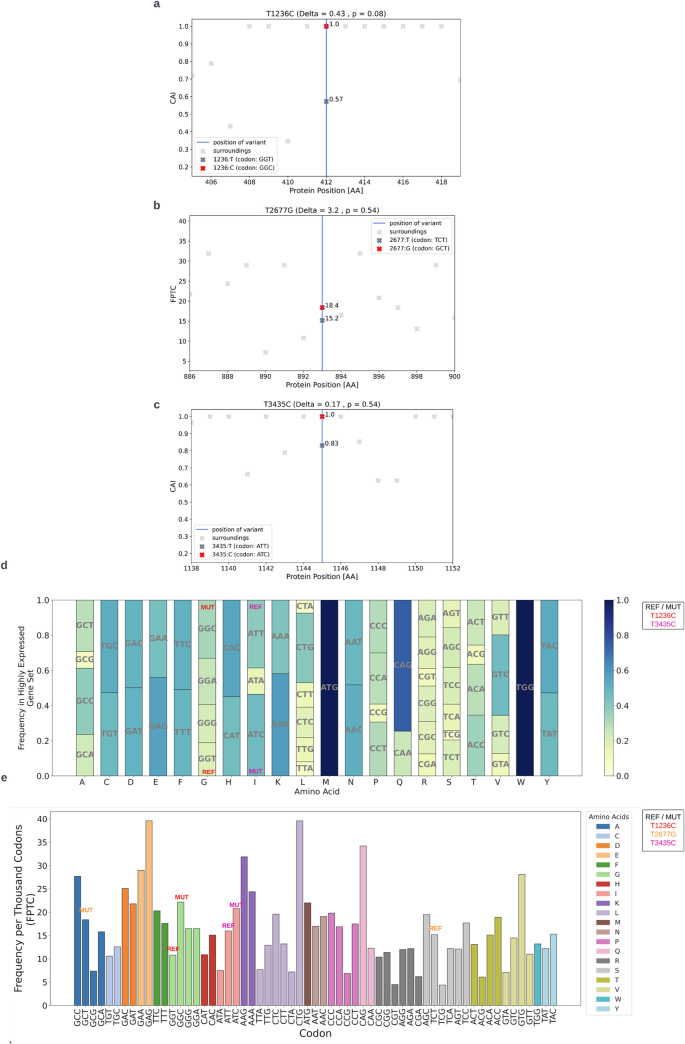
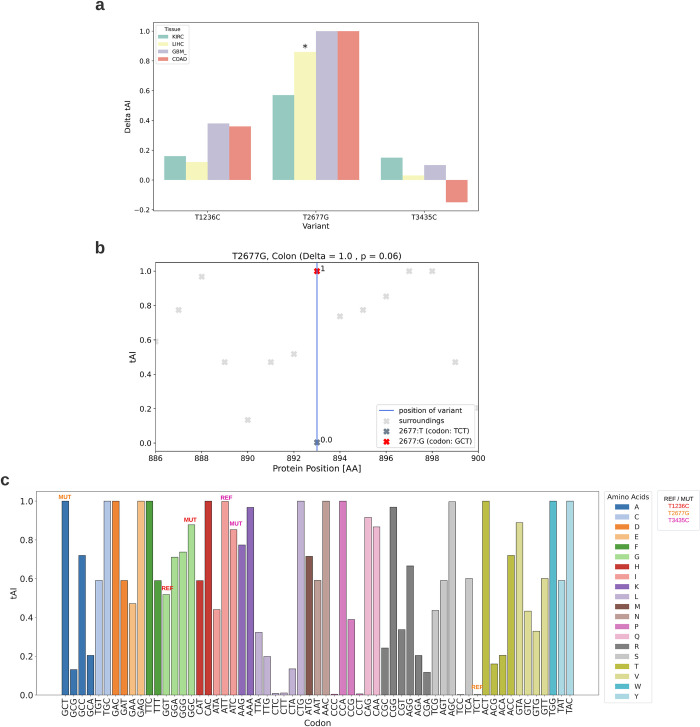
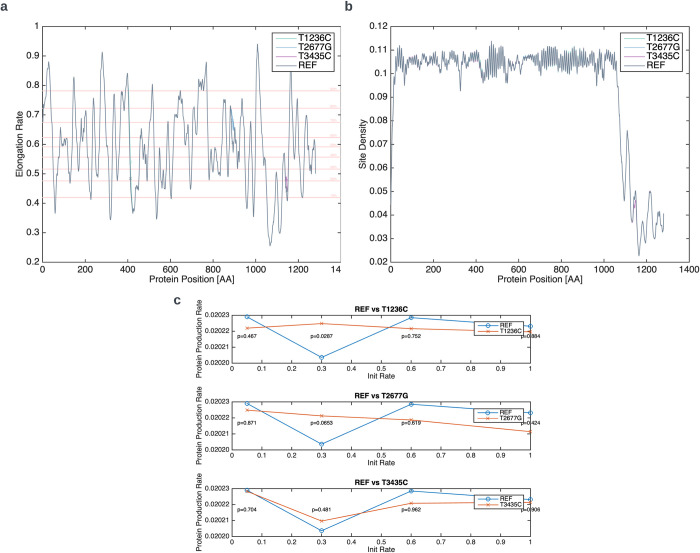
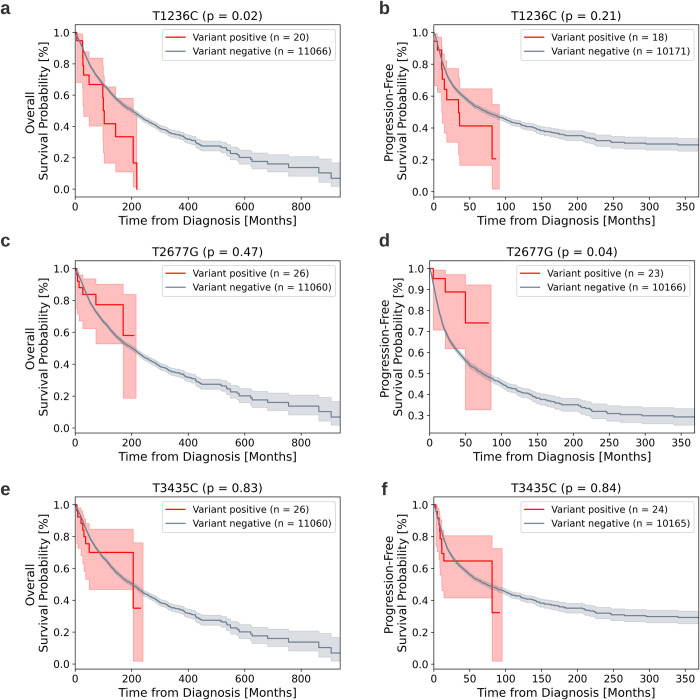
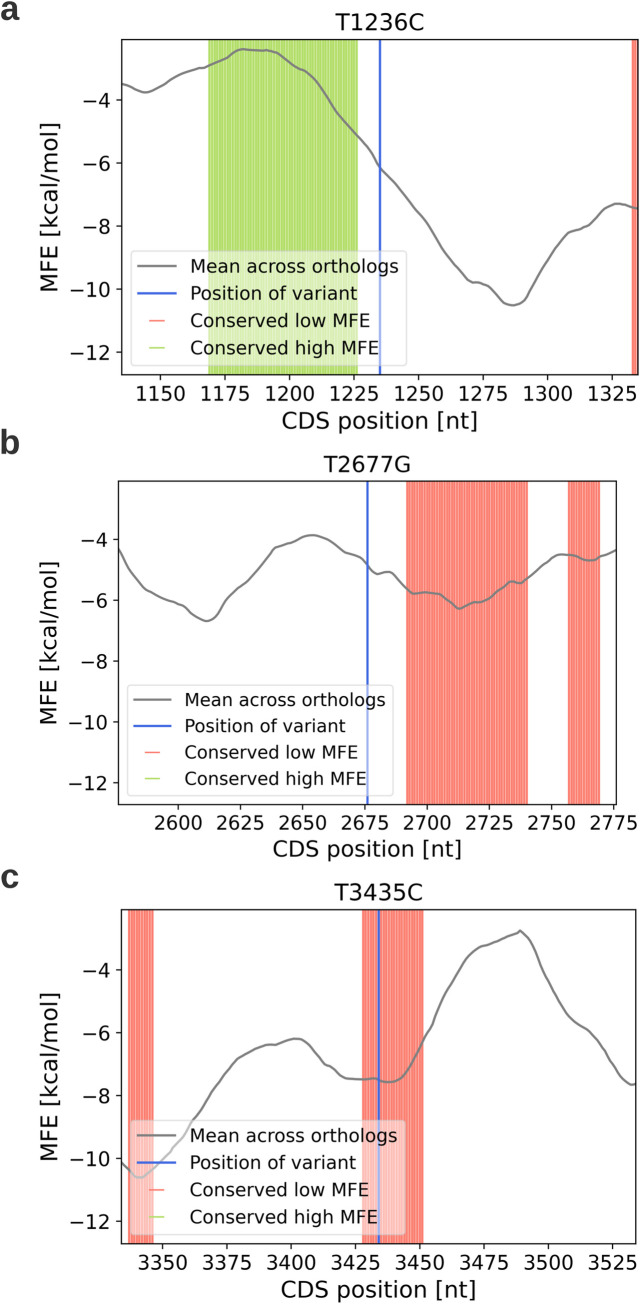
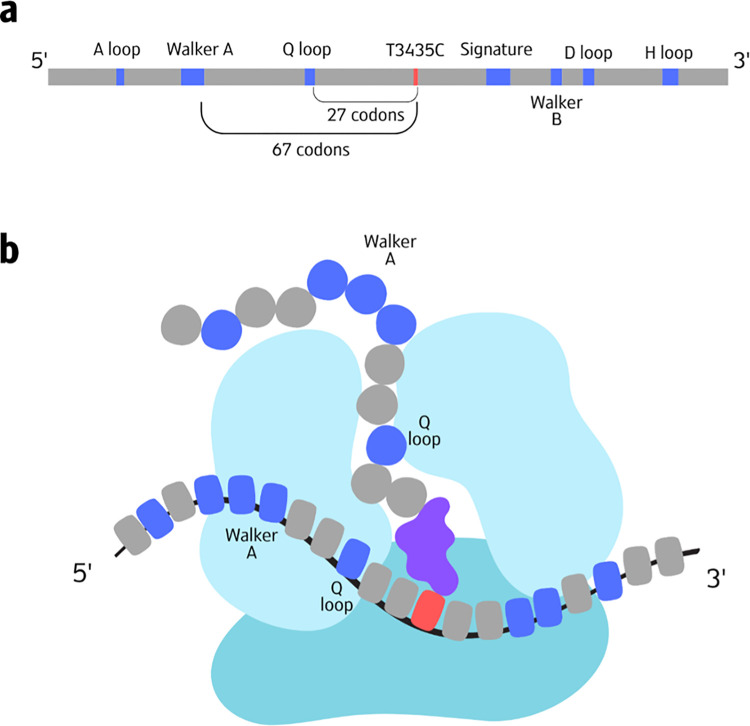
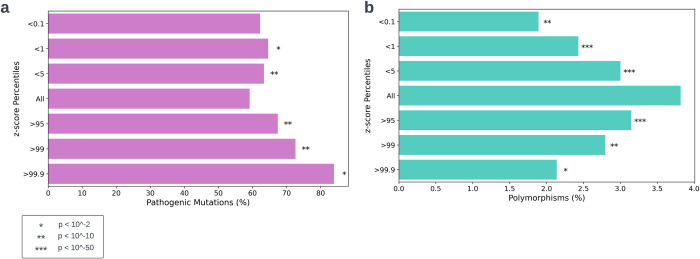
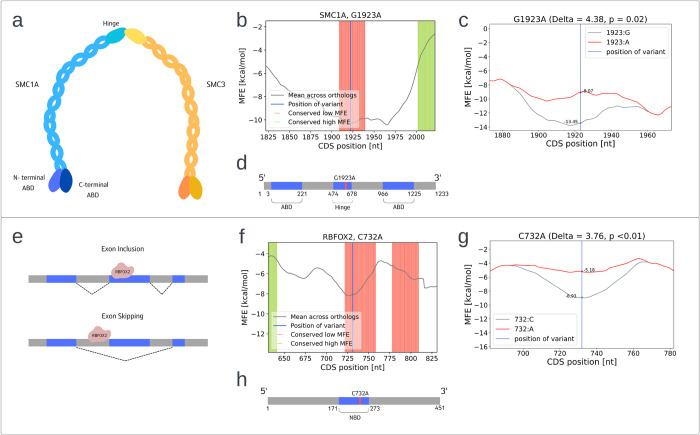
The P-glycoprotein efflux pump, encoded by the MDR1 gene, is an ATP-driven transporter capable of expelling a diverse array of compounds from cells. Overexpression of this protein is implicated in the multi-drug resistant phenotype observed in various cancers. Numerous studies have attempted to decipher the impact of genetic variants within MDR1 on P-glycoprotein expression, functional activity, and clinical outcomes in cancer patients. Among these, three specific single nucleotide polymorphisms-T1236C, T2677G, and T3435C - have been the focus of extensive research efforts, primarily through in vitro cell line models and clinical cohort analyses. However, the findings from these studies have been remarkably contradictory. In this study, we employ a computational, data-driven approach to systematically evaluate the effects of these three variants on principal stages of the gene expression process. Leveraging current knowledge of gene regulatory mechanisms, we elucidate potential mechanisms by which these variants could modulate P-glycoprotein levels and function. Our findings suggest that all three variants significantly change the mRNA folding in their vicinity. This change in mRNA structure is predicted to increase local translation elongation rates, but not to change the protein expression. Nonetheless, the increased translation rate near T3435C is predicted to affect the protein's co-translational folding trajectory in the region of the second ATP binding domain. This potentially impacts P-glycoprotein conformation and function. Our study demonstrates the value of computational approaches in elucidating the functional consequences of genetic variants. This framework provides new insights into the molecular mechanisms of MDR1 variants and their potential impact on cancer prognosis and treatment resistance. Furthermore, we introduce an approach which can be systematically applied to identify mutations potentially affecting mRNA folding in pathology. We demonstrate the utility of this approach on both ClinVar and TCGA and identify hundreds of disease related variants that modify mRNA folding at essential positions.
Copyright: © 2024 Gutman, Tuller. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


Similar articles
-
Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition.Xenobiotica. 2008 Jul;38(7-8):802-32. doi: 10.1080/00498250701867889. Xenobiotica. 2008. PMID: 18668431 Review.
-
Studies of human MDR1-MDR2 chimeras demonstrate the functional exchangeability of a major transmembrane segment of the multidrug transporter and phosphatidylcholine flippase.Mol Cell Biol. 1999 Feb;19(2):1450-9. doi: 10.1128/MCB.19.2.1450. Mol Cell Biol. 1999. PMID: 9891078 Free PMC article.
-
MDR1 and MDR3 genes and drug resistance to cisplatin of ovarian cancer cells.J Huazhong Univ Sci Technolog Med Sci. 2007 Dec;27(6):721-4. doi: 10.1007/s11596-007-0627-7. J Huazhong Univ Sci Technolog Med Sci. 2007. PMID: 18231753
-
MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells.Mol Pharmacol. 2013 Feb;83(2):399-405. doi: 10.1124/mol.112.081844. Epub 2012 Nov 19. Mol Pharmacol. 2013. PMID: 23166305
-
The role of MDR1 genetic polymorphisms in interindividual variability in P-glycoprotein expression and function.Curr Drug Metab. 2004 Feb;5(1):11-9. doi: 10.2174/1389200043489108. Curr Drug Metab. 2004. PMID: 14965248 Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
