

RNA gain-of-function mechanisms in short tandem repeat diseases
- PMID: 39725460
- PMCID: PMC11874975
- DOI: 10.1261/rna.080277.124
RNA gain-of-function mechanisms in short tandem repeat diseases
Abstract
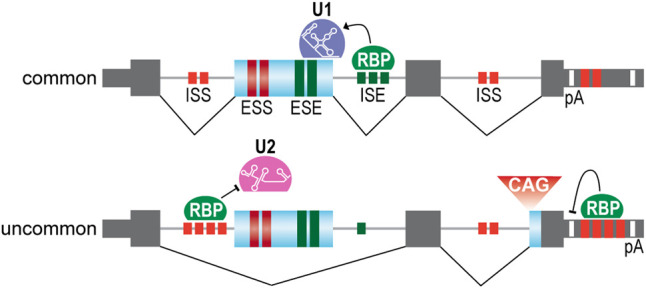
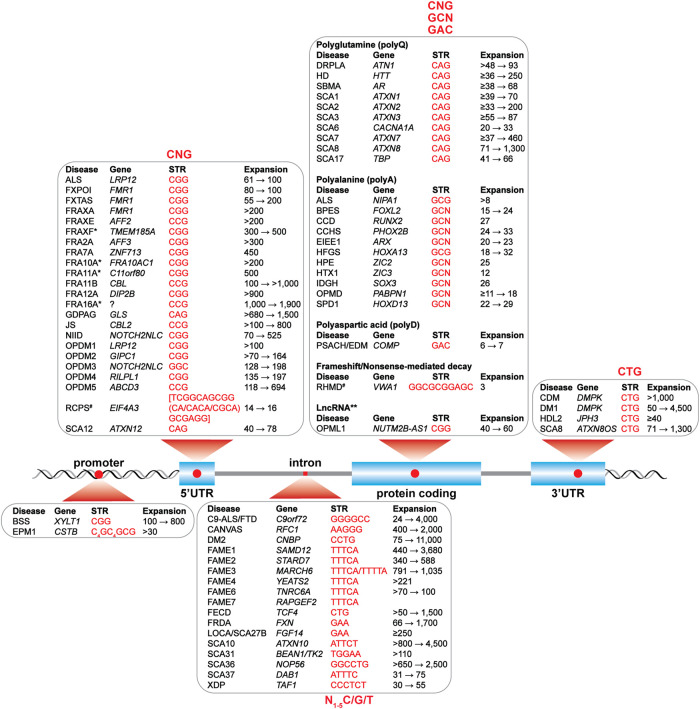
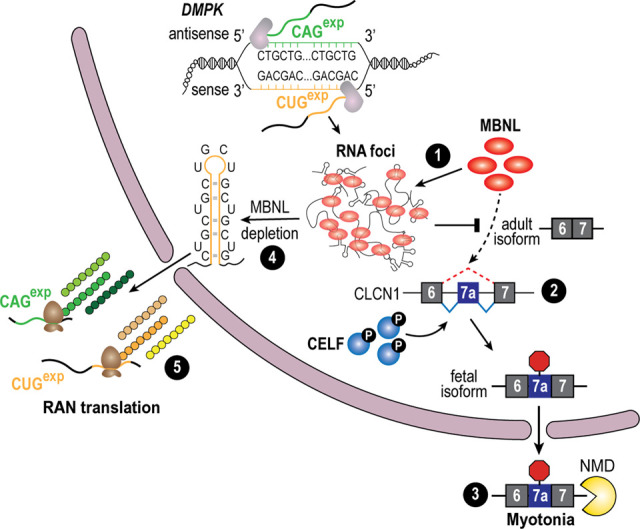
As adaptors, catalysts, guides, messengers, scaffolds, and structural components, RNAs perform an impressive array of cellular regulatory functions often by recruiting RNA-binding proteins (RBPs) to form ribonucleoprotein complexes (RNPs). While this RNA-RBP interaction network allows precise RNP assembly and the subsequent structural dynamics required for normal functions, RNA motif mutations may trigger the formation of aberrant RNP structures that lead to cell dysfunction and disease. Here, we provide our perspective on one type of RNA motif mutation, RNA gain-of-function mutations associated with the abnormal expansion of short tandem repeats (STRs) that underlie multiple developmental and degenerative diseases. We first discuss our current understanding of normal polymorphic STR functions in RNA processing and localization followed by an assessment of the pathogenic roles of STR expansions in the neuromuscular disease myotonic dystrophy. We also highlight ongoing questions and controversies focused on STR-based insights into the regulation of nuclear RNA processing and export as well as the relevance of the RNA gain-of-function pathomechanism for other STR expansion disorders in both coding and noncoding genes.
Keywords: RNA; anticipation; neurological disease; neuromuscular disease; short tandem repeat.
© 2025 Davenport and Swanson; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
Similar articles
-
Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy.Int J Mol Sci. 2019 Jul 9;20(13):3365. doi: 10.3390/ijms20133365. Int J Mol Sci. 2019. PMID: 31323950 Free PMC article. Review.
-
RNA-protein interactions in unstable microsatellite diseases.Brain Res. 2014 Oct 10;1584:3-14. doi: 10.1016/j.brainres.2014.03.039. Epub 2014 Apr 4. Brain Res. 2014. PMID: 24709120 Free PMC article. Review.
-
Cell-type-specific dysregulation of RNA alternative splicing in short tandem repeat mouse knockin models of myotonic dystrophy.Genes Dev. 2019 Dec 1;33(23-24):1635-1640. doi: 10.1101/gad.328963.119. Epub 2019 Oct 17. Genes Dev. 2019. PMID: 31624084 Free PMC article.
-
RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity.Brain Res. 2012 Jun 26;1462:100-11. doi: 10.1016/j.brainres.2012.02.030. Epub 2012 Feb 22. Brain Res. 2012. PMID: 22405728 Free PMC article. Review.
-
Gain of RNA function in pathological cases: Focus on myotonic dystrophy.Biochimie. 2011 Nov;93(11):2006-12. doi: 10.1016/j.biochi.2011.06.028. Epub 2011 Jul 13. Biochimie. 2011. PMID: 21763392 Review.
Cited by
-
Dysregulated RNA-binding proteins and alternative splicing: Emerging roles in autism spectrum disorder.Mol Cells. 2025 Aug;48(8):100237. doi: 10.1016/j.mocell.2025.100237. Epub 2025 Jun 3. Mol Cells. 2025. PMID: 40472970 Free PMC article. Review.
References
-
- Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, Collins RL, Domingo A, Currall B, Stortchevoi A, et al. 2018. Dissecting the causal mechanism of X-linked dystonia-parkinsonism by integrating genome and transcriptome assembly. Cell 172: 897–909.e21. 10.1016/j.cell.2018.02.011 - DOI - PMC - PubMed
-
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. 2014. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56: 311–322. 10.1016/j.molcel.2014.08.027 - DOI - PMC - PubMed
-
- Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, Dhakal J, Ton ML, Cheng A, Russo CT, et al. 2017. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci 114: E11020–E11028. 10.1073/pnas.1712526114 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials