

Endothelial CD38-induced endothelial-to-mesenchymal transition is a pivotal driver in pulmonary fibrosis
- PMID: 39725783
- PMCID: PMC11671674
- DOI: 10.1007/s00018-024-05548-x
Endothelial CD38-induced endothelial-to-mesenchymal transition is a pivotal driver in pulmonary fibrosis
Abstract
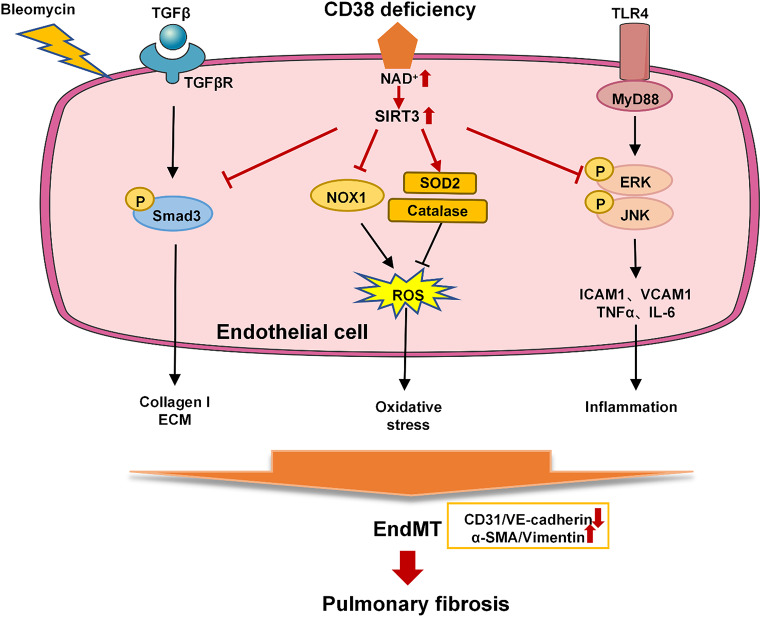
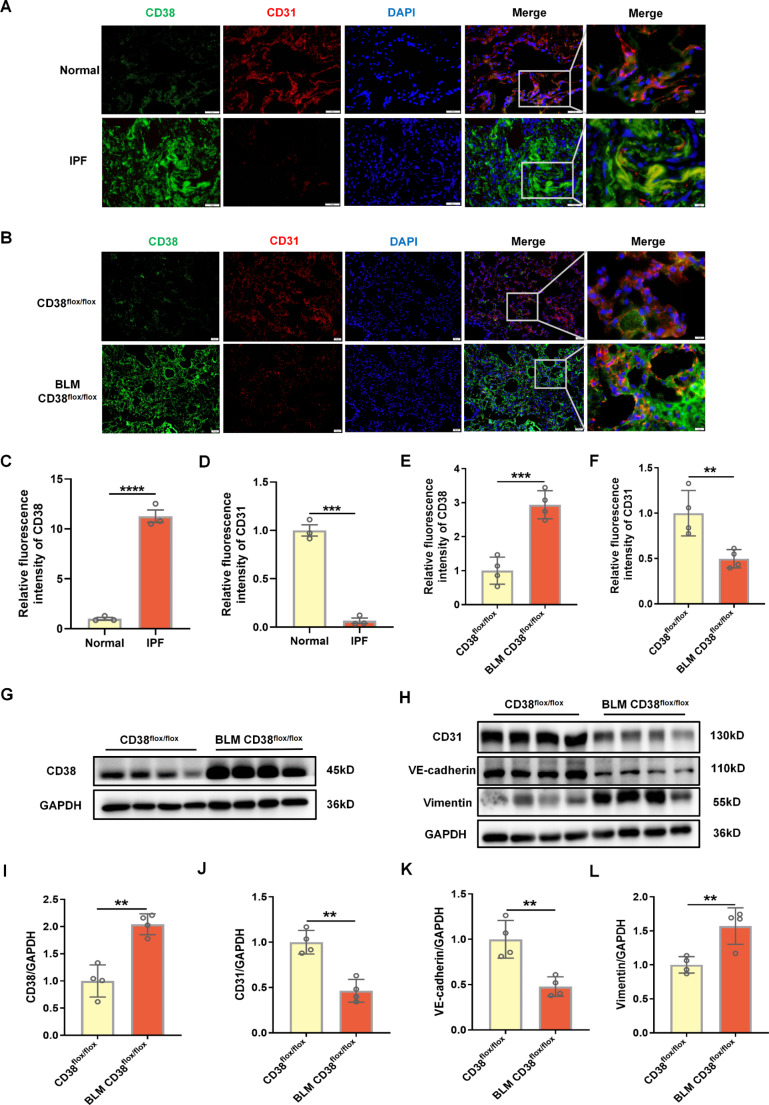
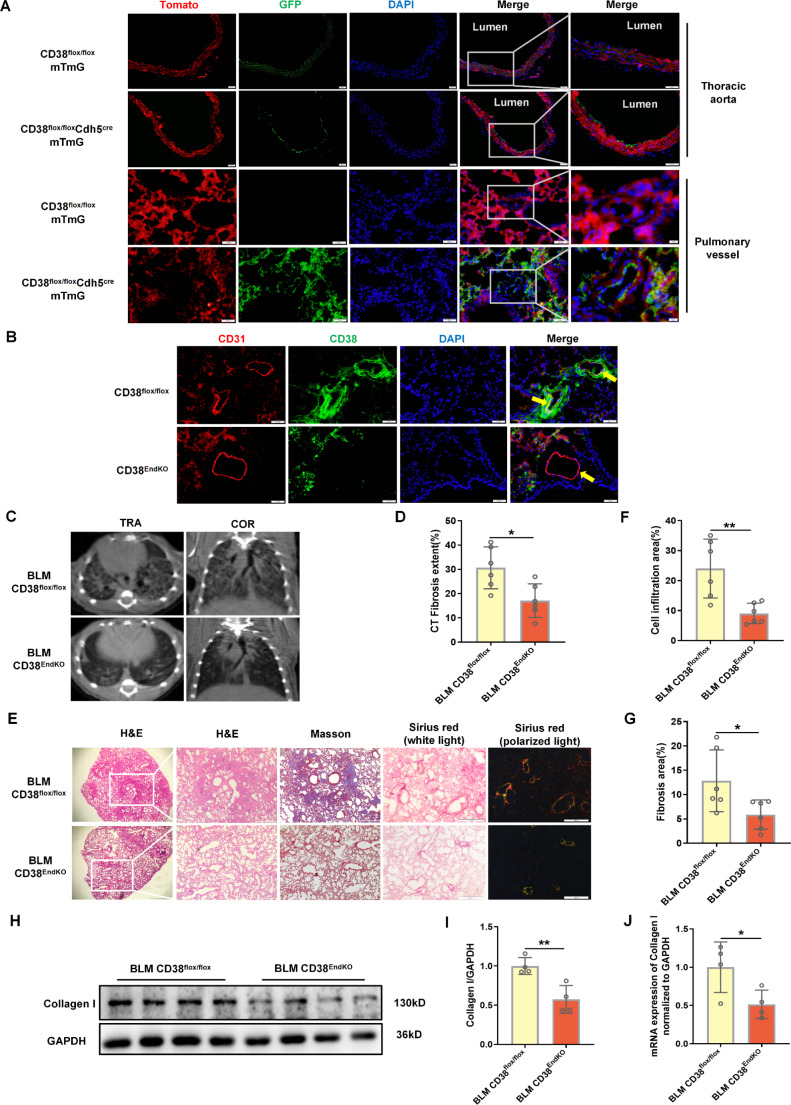
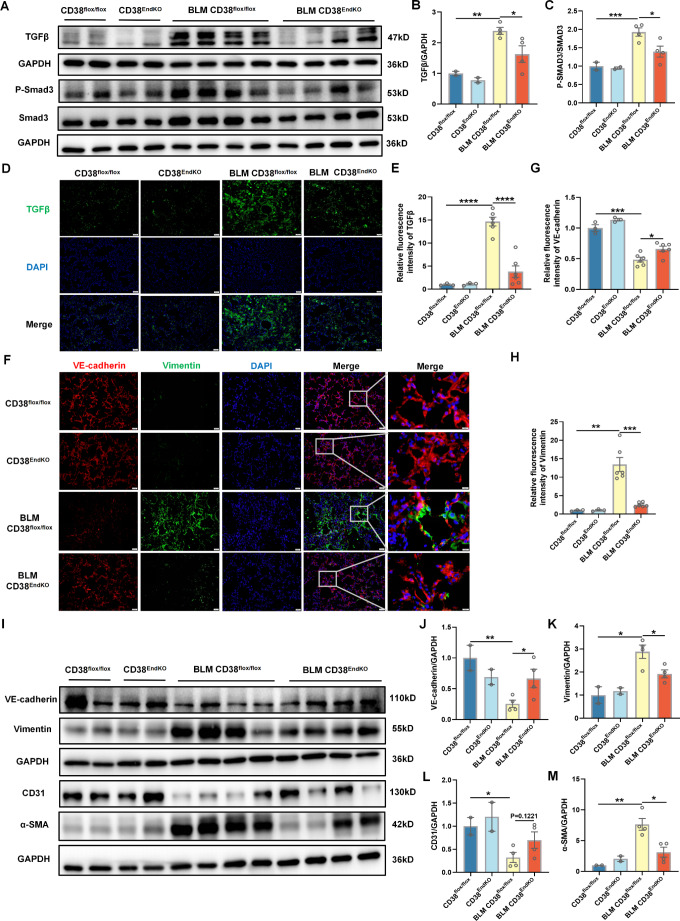
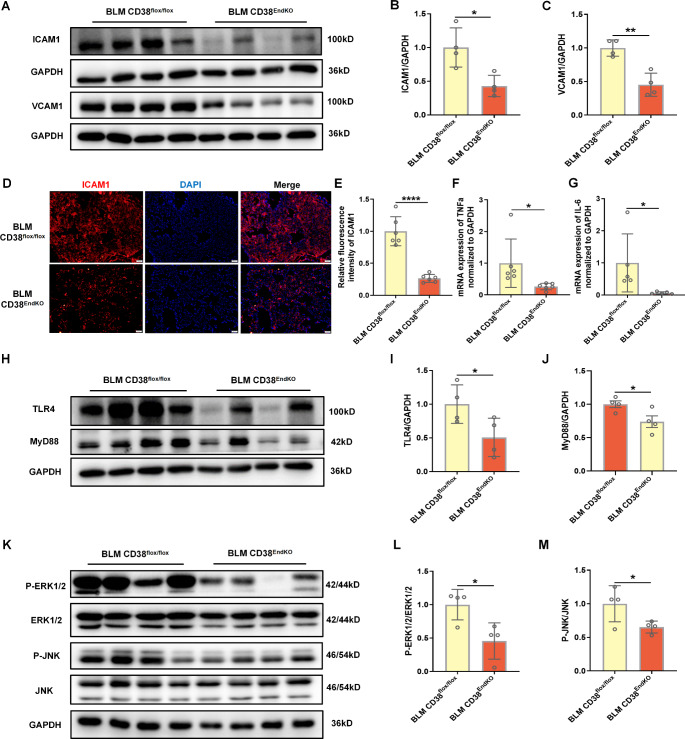
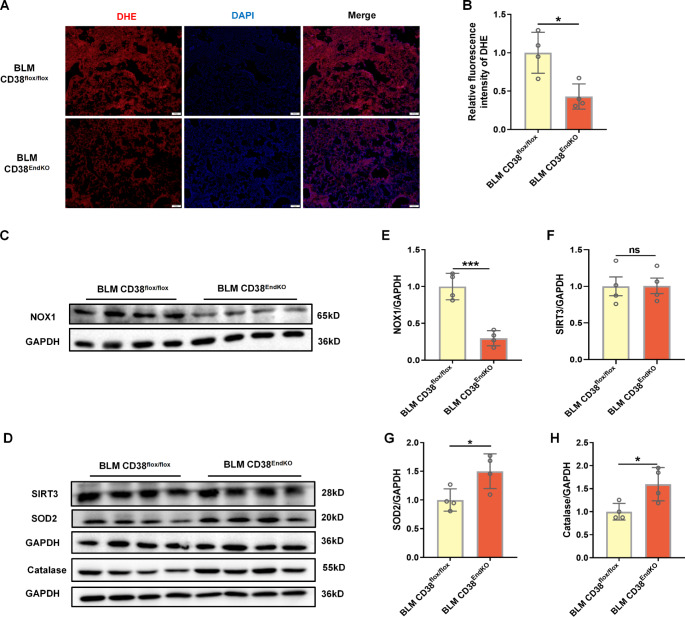
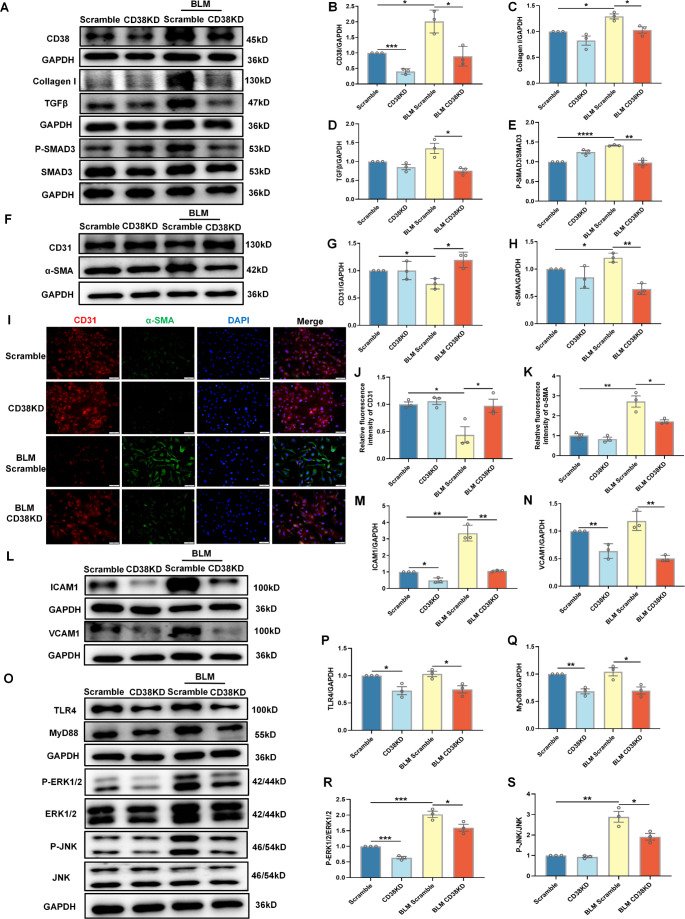
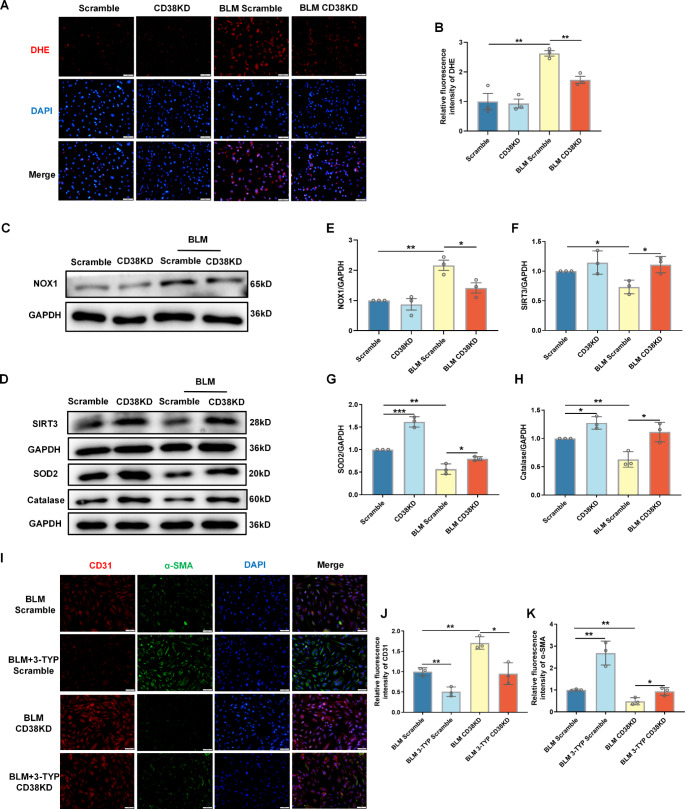
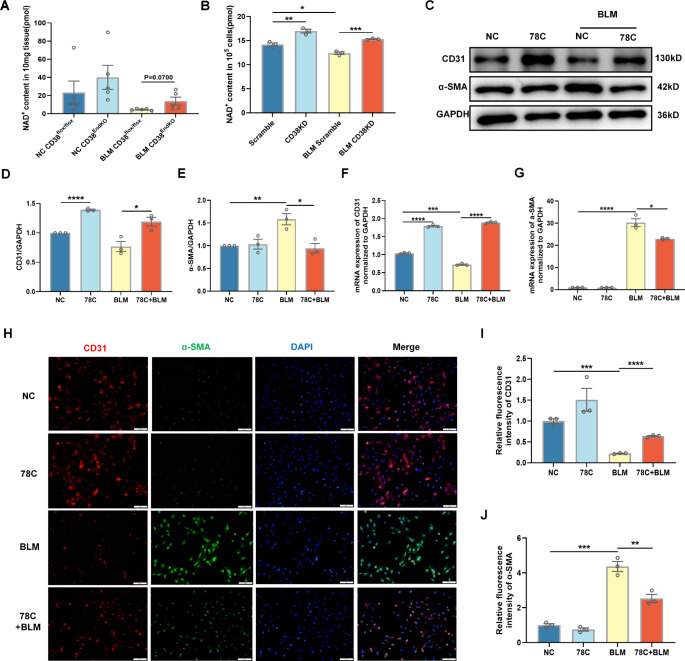
Idiopathic pulmonary fibrosis (IPF) is a prevalent interstitial lung disease with high mortality. CD38 is a main enzyme for intracellular nicotinamide adenine dinucleotide (NAD+) degradation in mammals. It has been reported that CD38 participated in pulmonary fibrosis through promoting alveolar epithelial cells senescence. However, the roles of endothelial CD38 in pulmonary fibrosis remain unknown. In the present study, we observed that the elevated expression of CD38 was related to endothelial-to-mesenchymal transition (EndMT) of lung tissues in IPF patients and bleomycin (BLM)-induced pulmonary fibrosis mice and also in human umbilical vein endothelial cells (HUVECs) treated with BLM. Micro-computed tomography (MCT) and histopathological staining showed that endothelial cell-specific CD38 knockout (CD38EndKO) remarkably attenuated BLM-induced pulmonary fibrosis. In addition, CD38EndKO significantly inhibited TGFβ-Smad3 pathway-mediated excessive extracellular matrix (ECM), reduced Toll-like receptor4-Myeloid differentiation factor88-Mitogen-activated protein kinases (TLR4-MyD88-MAPK) pathway-mediated endothelial inflammation and suppressed nicotinamide adenine dinucleotide phosphate oxidases1 (NOX1)-mediated oxidative stress. Furthermore, we demonstrated that 3-TYP, a SIRT3-specific inhibitor, markedly reversed the protective effect of HUVECsCD38KD cells and 78 C, a CD38-specific inhibitor, on BLM-induced EndMT in HUVECs. Therefore, we concluded that CD38EndKO significantly ameliorated BLM-induced pulmonary fibrosis through inhibiting ECM, endothelial inflammation and oxidative stress, further alleviating EndMT in mice. Our findings suggest that endothelial CD38 may be a new therapeutic target for the prevention and treatment of pulmonary fibrosis clinically.
Keywords: CD38; Endothelial-to-mesenchymal transition; Inflammation; Oxidative stress; Pulmonary fibrosis.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: All the experimental procedures were approved by Nanchang University Institutional Animal Research Committee (Date: 20230822/No: 23 × 007) and were carried out in accordance with Jiangxi Province Laboratory Animal Care Guidelines for the use of animals in research. Consent to participate: Not applicable. Consent for publication: All authors have read the manuscript and agree with submission for publication. Competing interests: The authors have no relevant financial or non-financial interests to disclose.
Figures
Similar articles
-
Tauroursodeoxycholic acid alleviates pulmonary endoplasmic reticulum stress and epithelial-mesenchymal transition in bleomycin-induced lung fibrosis.BMC Pulm Med. 2021 May 5;21(1):149. doi: 10.1186/s12890-021-01514-6. BMC Pulm Med. 2021. PMID: 33952237 Free PMC article.
-
The role of quercetin in ameliorating bleomycin-induced pulmonary fibrosis: insights into autophagy and the SIRT1/AMPK signaling pathway.Mol Biol Rep. 2024 Jul 13;51(1):795. doi: 10.1007/s11033-024-09752-7. Mol Biol Rep. 2024. PMID: 39001907
-
Urine-Derived Stem Cells Reverse Bleomycin‑Induced Experimental Pulmonary Fibrosis by Inhibition of the TGF-β1-Smad2/3 Pathway.Cytotherapy. 2024 Oct;26(10):1236-1244. doi: 10.1016/j.jcyt.2024.05.015. Epub 2024 May 17. Cytotherapy. 2024. PMID: 38852093
-
S1PR1 attenuates pulmonary fibrosis by inhibiting EndMT and improving endothelial barrier function.Pulm Pharmacol Ther. 2023 Aug;81:102228. doi: 10.1016/j.pupt.2023.102228. Epub 2023 Jun 8. Pulm Pharmacol Ther. 2023. PMID: 37295666
-
Biological and pharmacological roles of pyroptosis in pulmonary inflammation and fibrosis: recent advances and future directions.Cell Commun Signal. 2024 Dec 5;22(1):586. doi: 10.1186/s12964-024-01966-3. Cell Commun Signal. 2024. PMID: 39639365 Free PMC article. Review.
References
-
- Wang X, Wang Y, Rong S, Ma H (2014) [Early treatment with hepatocyte growth factor improves pulmonary artery and right ventricular remodeling in rats with pulmonary artery hypertension by modulating cytokines expression]. Zhonghua Jie He He Hu Xi Za Zhi 37:427–432 - PubMed
-
- Spagnolo P, Cottin V (2017) Genetics of idiopathic pulmonary fibrosis: from mechanistic pathways to personalised medicine. J Med Genet 54:93–99 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
