

Machine learning-optimized targeted detection of alternative splicing
- PMID: 39727154
- PMCID: PMC11797022
- DOI: 10.1093/nar/gkae1260
Machine learning-optimized targeted detection of alternative splicing
Abstract
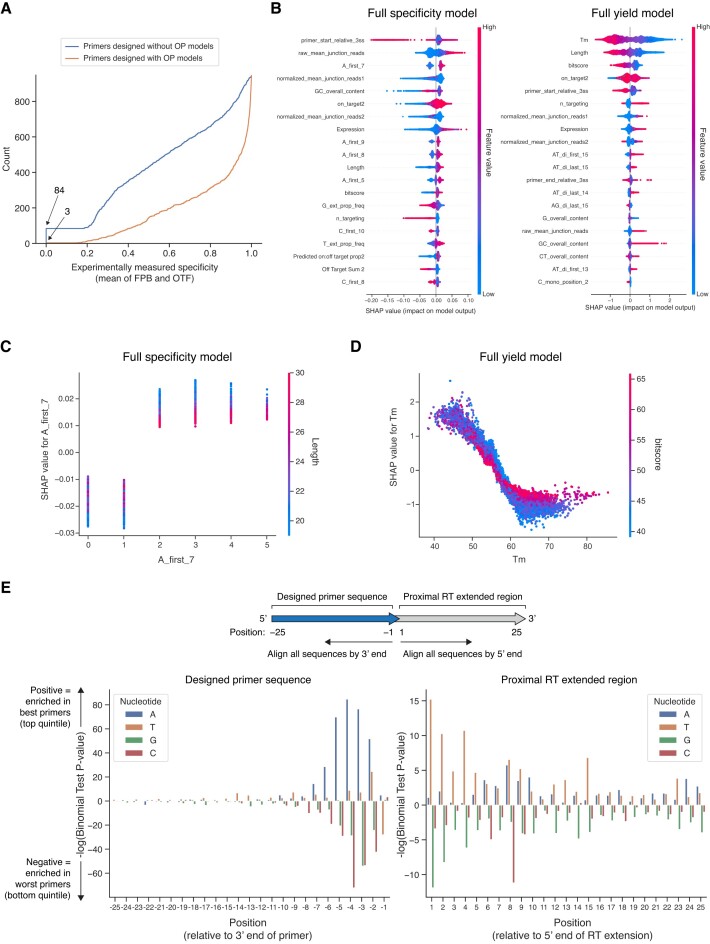
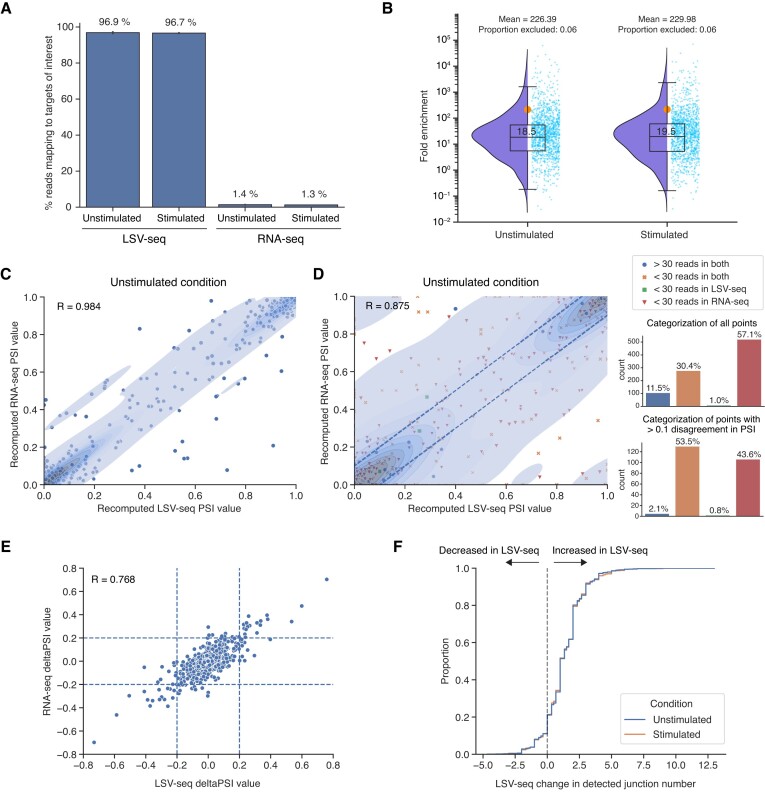
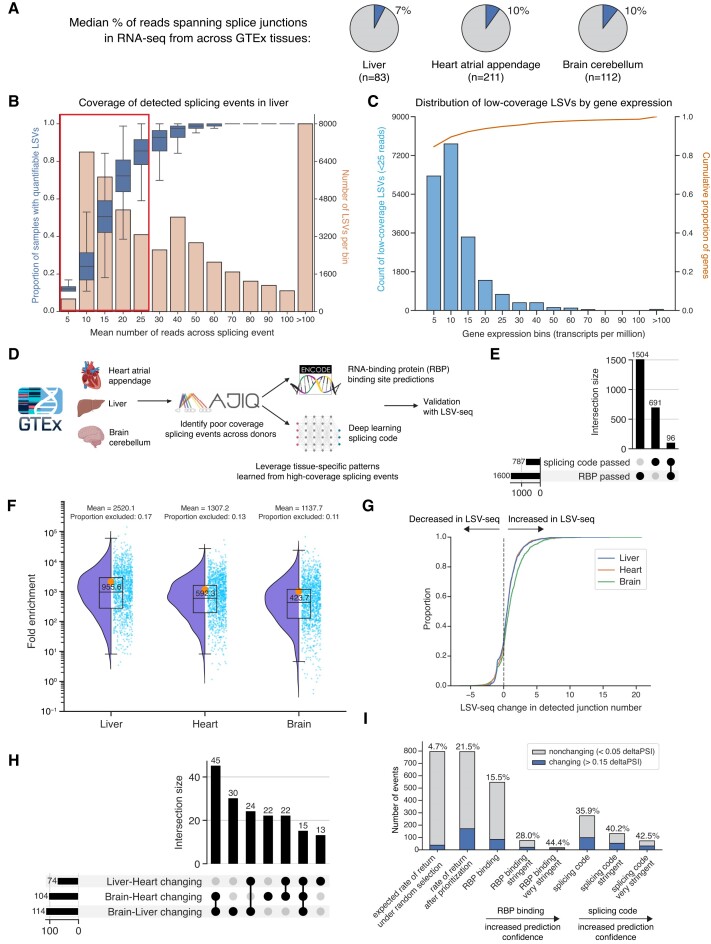
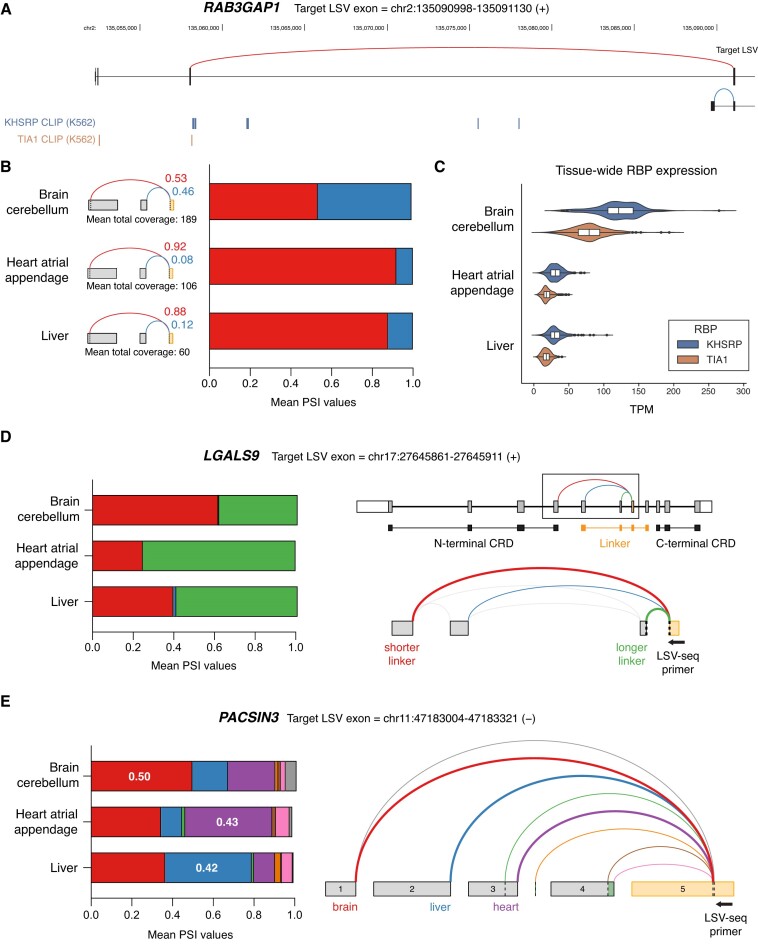
RNA sequencing (RNA-seq) is widely adopted for transcriptome analysis but has inherent biases that hinder the comprehensive detection and quantification of alternative splicing. To address this, we present an efficient targeted RNA-seq method that greatly enriches for splicing-informative junction-spanning reads. Local splicing variation sequencing (LSV-seq) utilizes multiplexed reverse transcription from highly scalable pools of primers anchored near splicing events of interest. Primers are designed using Optimal Prime, a novel machine learning algorithm trained on the performance of thousands of primer sequences. In experimental benchmarks, LSV-seq achieves high on-target capture rates and concordance with RNA-seq, while requiring significantly lower sequencing depth. Leveraging deep learning splicing code predictions, we used LSV-seq to target events with low coverage in GTEx RNA-seq data and newly discover hundreds of tissue-specific splicing events. Our results demonstrate the ability of LSV-seq to quantify splicing of events of interest at high-throughput and with exceptional sensitivity.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures



Update of
-
Machine learning-optimized targeted detection of alternative splicing.bioRxiv [Preprint]. 2024 Sep 24:2024.09.20.614162. doi: 10.1101/2024.09.20.614162. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkae1260. doi: 10.1093/nar/gkae1260. PMID: 39386495 Free PMC article. Updated. Preprint.
Similar articles
-
Machine learning-optimized targeted detection of alternative splicing.bioRxiv [Preprint]. 2024 Sep 24:2024.09.20.614162. doi: 10.1101/2024.09.20.614162. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkae1260. doi: 10.1093/nar/gkae1260. PMID: 39386495 Free PMC article. Updated. Preprint.
-
rMATS-turbo: an efficient and flexible computational tool for alternative splicing analysis of large-scale RNA-seq data.Nat Protoc. 2024 Apr;19(4):1083-1104. doi: 10.1038/s41596-023-00944-2. Epub 2024 Feb 23. Nat Protoc. 2024. PMID: 38396040 Review.
-
Incorporating exon-exon junction reads enhances differential splicing detection.BMC Bioinformatics. 2025 Jul 24;26(1):193. doi: 10.1186/s12859-025-06210-4. BMC Bioinformatics. 2025. PMID: 40707876 Free PMC article.
-
Multimodal zero-shot learning of previously unseen epitranscriptomes from RNA-seq data.Brief Bioinform. 2025 Jul 2;26(4):bbaf332. doi: 10.1093/bib/bbaf332. Brief Bioinform. 2025. PMID: 40632498 Free PMC article.
-
Sequential versus standard triple first-line therapy for Helicobacter pylori eradication.Cochrane Database Syst Rev. 2016 Jun 28;2016(6):CD009034. doi: 10.1002/14651858.CD009034.pub2. Cochrane Database Syst Rev. 2016. PMID: 27351542 Free PMC article.
Cited by
-
Generative modeling for RNA splicing predictions and design.bioRxiv [Preprint]. 2025 Jan 24:2025.01.20.633986. doi: 10.1101/2025.01.20.633986. bioRxiv. 2025. PMID: 39896553 Free PMC article. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
