

Biomimetic [MFe3S4]3+ Cubanes (M = V/Mo) as Catalysts for a Fischer-Tropsch-like Hydrocarbon Synthesis─A Computational Study
- PMID: 39727298
- PMCID: PMC11734119
- DOI: 10.1021/acs.inorgchem.4c04995
Biomimetic [MFe3S4]3+ Cubanes (M = V/Mo) as Catalysts for a Fischer-Tropsch-like Hydrocarbon Synthesis─A Computational Study
Abstract
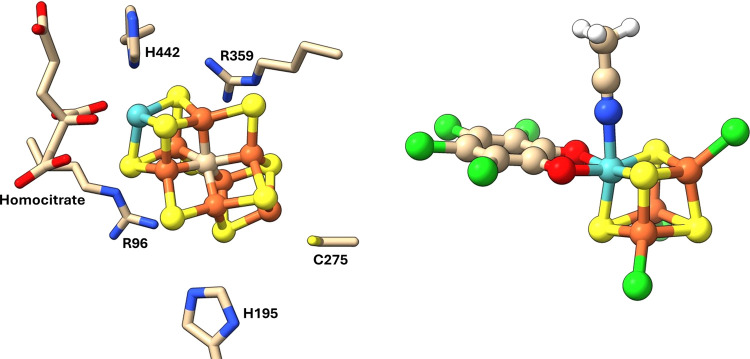
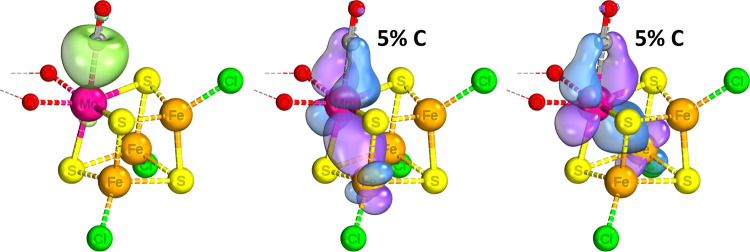
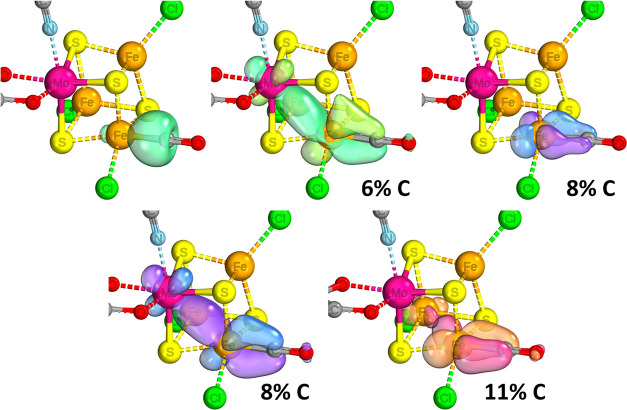
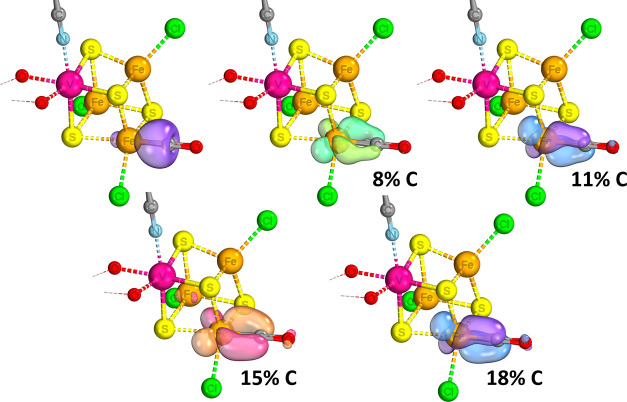
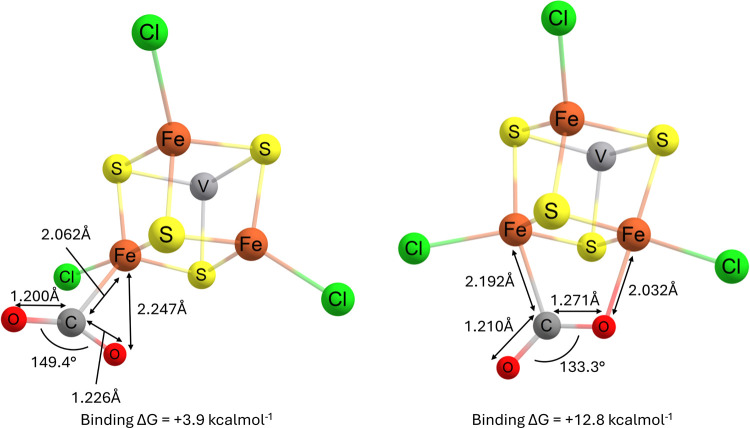
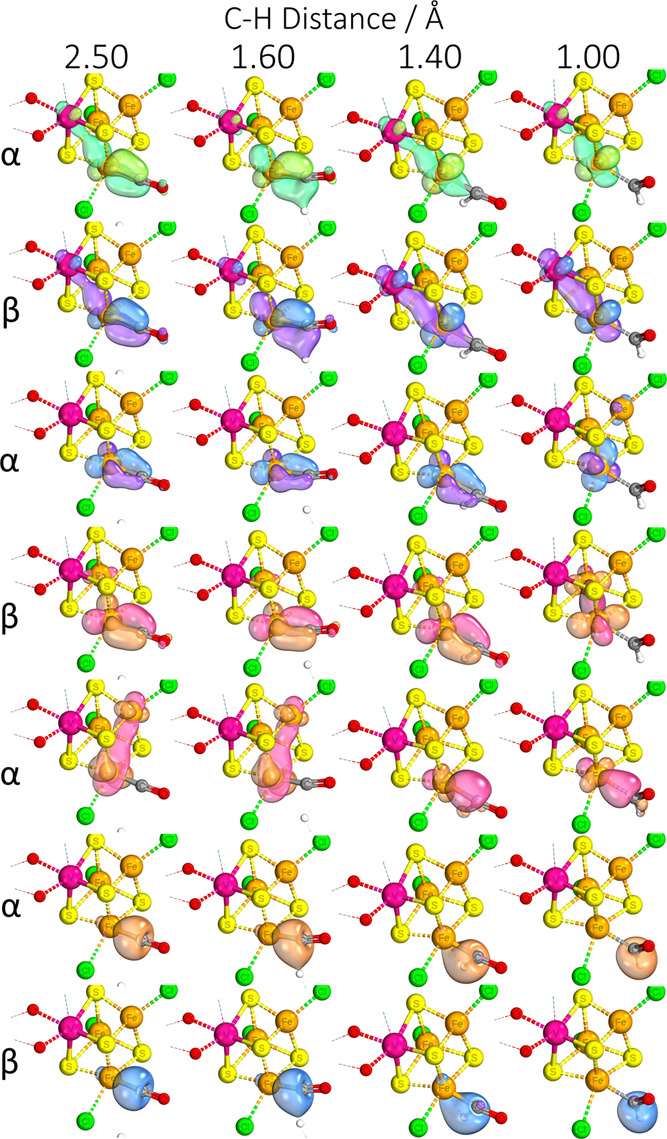
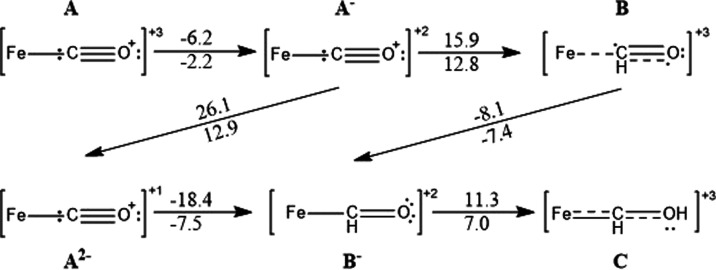
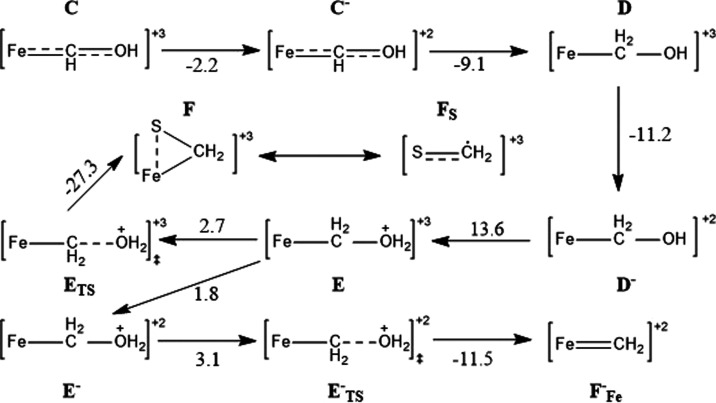
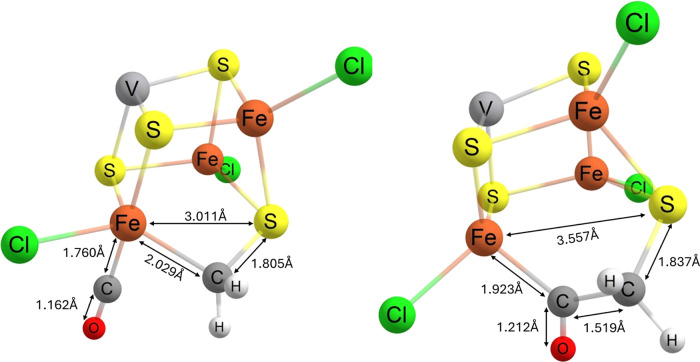
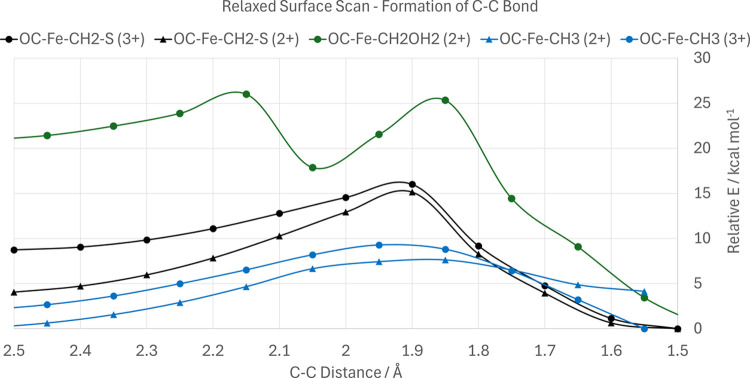
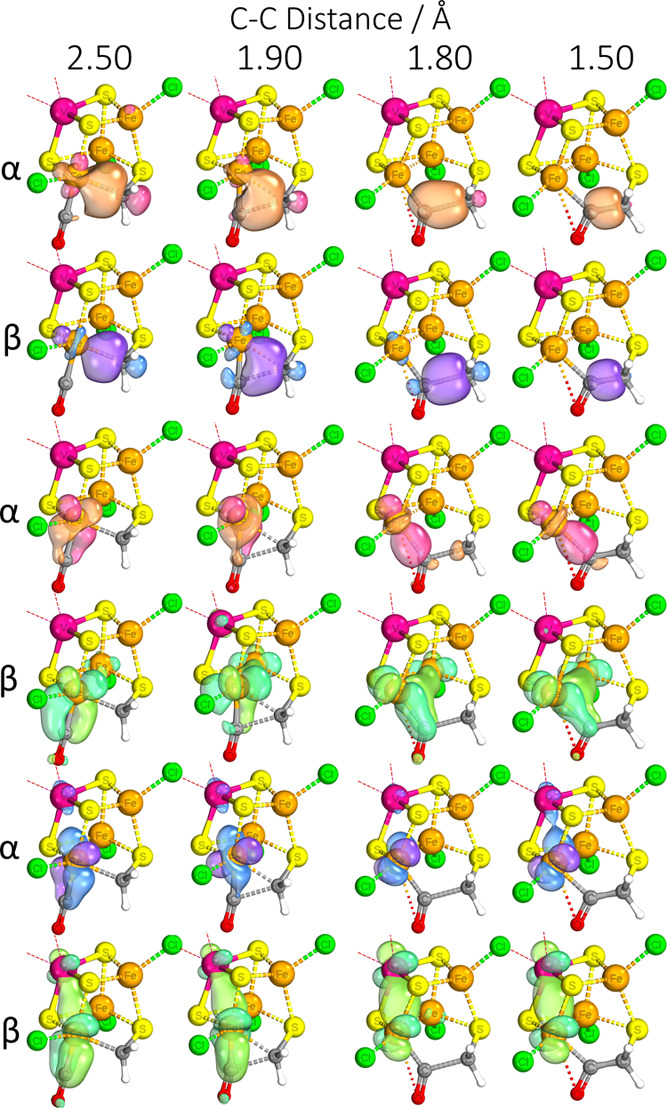
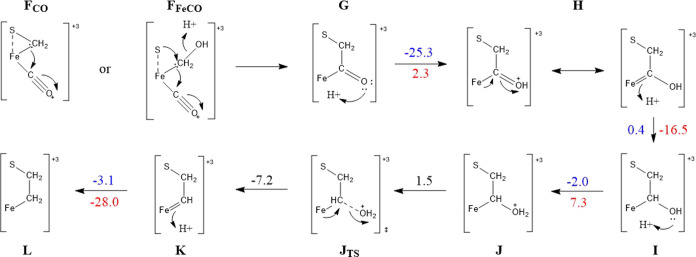
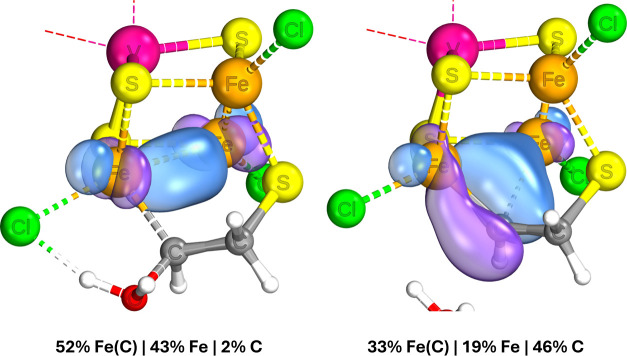
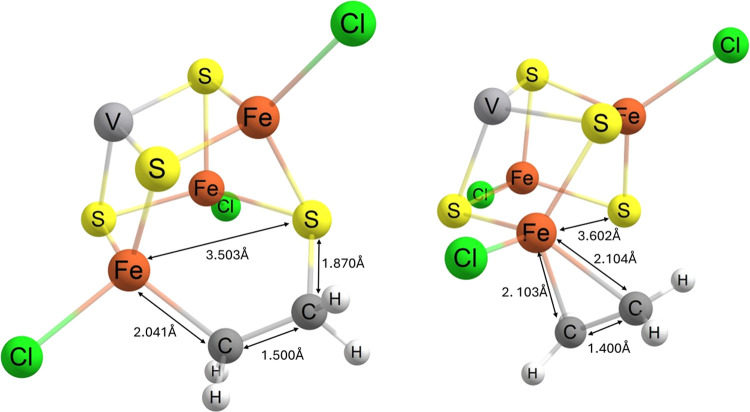
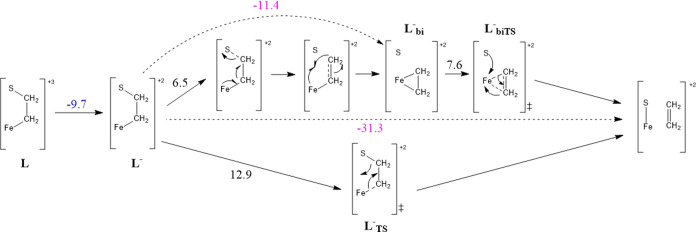
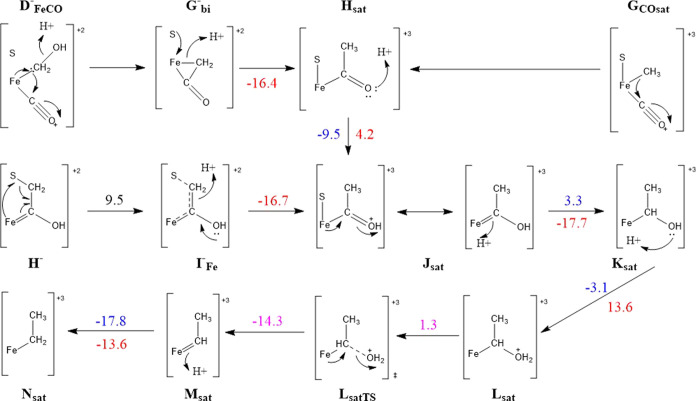
Nitrogenase is the enzyme primarily responsible for reducing atmospheric nitrogen to ammonia. There are three general forms of nitrogenase based on the metal ion present in the cofactor binding site, namely, molybdenum-dependent nitrogenases with the iron-molybdenum cofactor (FeMoco), the vanadium-dependent nitrogenases with FeVco, and the iron-only nitrogenases. It has been shown that the vanadium-dependent nitrogenases tend to have a lesser efficacy in reducing dinitrogen but a higher efficacy in binding and reducing carbon monoxide. In biomimetic chemistry, [MFe3S4] (M = Mo/V) cubanes have been synthesized, studied, and shown to be promising mimics of some of the geometric and electronic properties of the nitrogenase cofactors. In this work, a density functional theory (DFT) study is presented on Fischer-Tropsch catalysis by these cubane complexes by studying CO binding and reduction to hydrocarbons. Our work implies that molybdenum has stronger binding interactions with the iron-sulfur framework of the cubane, which results in easier reduction of substrates like N2H4. However, this inhibits the binding and activation of CO, and hence, the molybdenum-containing complexes are less suitable for Fischer-Tropsch catalysis than vanadium-containing complexes.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

Similar articles
-
Computational Study on the Influence of Mo/V Centers on the Electronic Structure and Hydrazine Reduction Capability of [MFe3S4]3+/2+ Complexes.Inorg Chem. 2023 Oct 9;62(40):16401-16411. doi: 10.1021/acs.inorgchem.3c02072. Epub 2023 Sep 27. Inorg Chem. 2023. PMID: 37756478 Free PMC article.
-
A Comparative Analysis of the CO-Reducing Activities of MoFe Proteins Containing Mo- and V-Nitrogenase Cofactors.Chembiochem. 2018 Apr 4;19(7):649-653. doi: 10.1002/cbic.201800035. Epub 2018 Feb 16. Chembiochem. 2018. PMID: 29363247
-
Ligand metathesis as rational strategy for the synthesis of cubane-type heteroleptic iron-sulfur clusters relevant to the FeMo cofactor.Proc Natl Acad Sci U S A. 2018 May 15;115(20):5089-5092. doi: 10.1073/pnas.1801025115. Epub 2018 Apr 13. Proc Natl Acad Sci U S A. 2018. PMID: 29654147 Free PMC article.
-
Biosynthesis of the Metalloclusters of Nitrogenases.Annu Rev Biochem. 2016 Jun 2;85:455-83. doi: 10.1146/annurev-biochem-060614-034108. Epub 2016 Feb 1. Annu Rev Biochem. 2016. PMID: 26844394 Review.
-
Cleaving the n,n triple bond: the transformation of dinitrogen to ammonia by nitrogenases.Met Ions Life Sci. 2014;14:147-76. doi: 10.1007/978-94-017-9269-1_7. Met Ions Life Sci. 2014. PMID: 25416394 Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
