

iPSC-derived retinal pigment epithelium: an in vitro platform to reproduce key cellular phenotypes and pathophysiology of retinal degenerative diseases
- PMID: 39729520
- PMCID: PMC11954503
- DOI: 10.1093/stcltm/szae097
iPSC-derived retinal pigment epithelium: an in vitro platform to reproduce key cellular phenotypes and pathophysiology of retinal degenerative diseases
Abstract
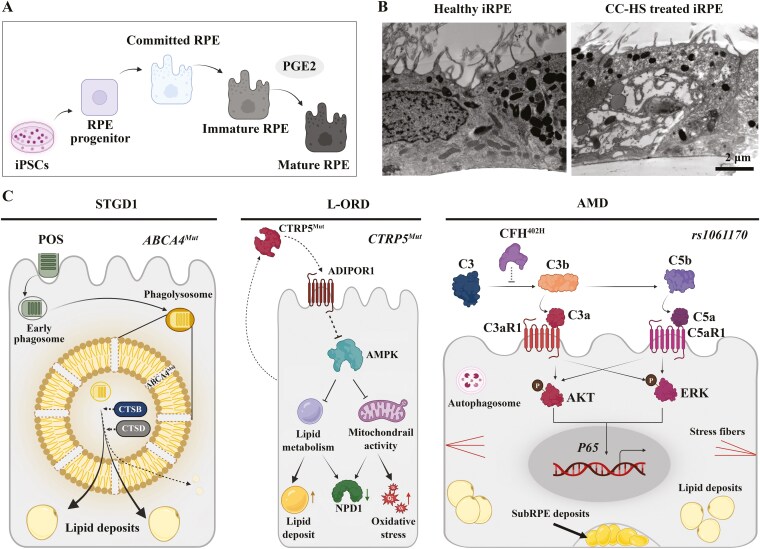
Retinal pigment epithelium (RPE) atrophy is a significant cause of human blindness worldwide, occurring in polygenic diseases such as age-related macular degeneration (AMD) and monogenic diseases such as Stargardt diseases (STGD1) and late-onset retinal degeneration (L-ORD). The patient-induced pluripotent stem cells (iPSCs)-derived RPE (iRPE) model exhibits many advantages in understanding the cellular basis of pathological mechanisms of RPE atrophy. The iRPE model is based on iPSC-derived functionally mature and polarized RPE cells that reproduce several features of native RPE cells, such as phagocytosis of photoreceptor outer segments (POS) and replenishment of visual pigment. When derived from patients, iRPE are able to recapitulate critical cellular phenotypes of retinal degenerative diseases, such as the drusen-like sub-RPE deposits in the L-ORD and AMD models; lipid droplets and cholesterol accumulation in the STGD1 and AMD models. The iRPE model has helped discover the unexpected role of RPE in understanding retinal degenerative diseases, such as a cell-autonomous function of ABCA4 in STGD1. The iRPE model has helped uncover the pathological mechanism of retinal degenerative diseases, including the roles of alternate complement cascades and oxidative stress in AMD pathophysiology, abnormal POS processing in STGD1 and L-ORD, and its association with lipid accumulation. These studies have helped better understand-the role of RPE in retinal degenerative diseases, and molecular mechanisms underlying RPE atrophy, and have provided a basis to discover therapeutics to target RPE-associated diseases.
Keywords: RPE atrophy; cell disease model; iPSCs-derived RPE; retinal degenerative diseases.
Published by Oxford University Press for the Infectious Diseases Society of America 2024.
Conflict of interest statement
The authors declared no potential conflicts of interest.
Figures
References
-
- Bharti K, Nguyen MT, Skuntz S, Bertuzzi S, Arnheiter H.. The other pigment cell: specification and development of the pigmented epithelium of the vertebrate eye. Pigment Cell Res. 2006;19:380-394. https://doi.org/10.1111/j.1600-0749.2006.00318.x - DOI - PMC - PubMed
-
- Ma X, Li H, Chen Y, et al.The transcription factor MITF in RPE function and dysfunction. Prog Retin Eye Res. 2019;73:100766. https://doi.org/10.1016/j.preteyeres.2019.06.002 - DOI - PubMed
-
- Ou J, Bharti K, Nodari A, Bertuzzi S, Arnheiter H.. Vax1/2 genes counteract Mitf-induced respecification of the retinal pigment epithelium. PLoS One. 2013;8:e59247. https://doi.org/10.1371/journal.pone.0059247 - DOI - PMC - PubMed
-
- Raviv S, Bharti K, Rencus-Lazar S, et al.PAX6 regulates melanogenesis in the retinal pigmented epithelium through feed-forward regulatory interactions with MITF. PLoS Genet. 2014;10:e1004360. https://doi.org/10.1371/journal.pgen.1004360 - DOI - PMC - PubMed
-
- Bharti K, Gasper M, Ou J, et al.A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet. 2012;8:e1002757. https://doi.org/10.1371/journal.pgen.1002757 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
