

Transcriptional analysis reveals the suppression of RAD51 and disruption of the homologous recombination pathway during PEDV infection in IPEC-J2 cells
- PMID: 39731192
- PMCID: PMC11681661
- DOI: 10.1186/s12985-024-02611-8
Transcriptional analysis reveals the suppression of RAD51 and disruption of the homologous recombination pathway during PEDV infection in IPEC-J2 cells
Abstract
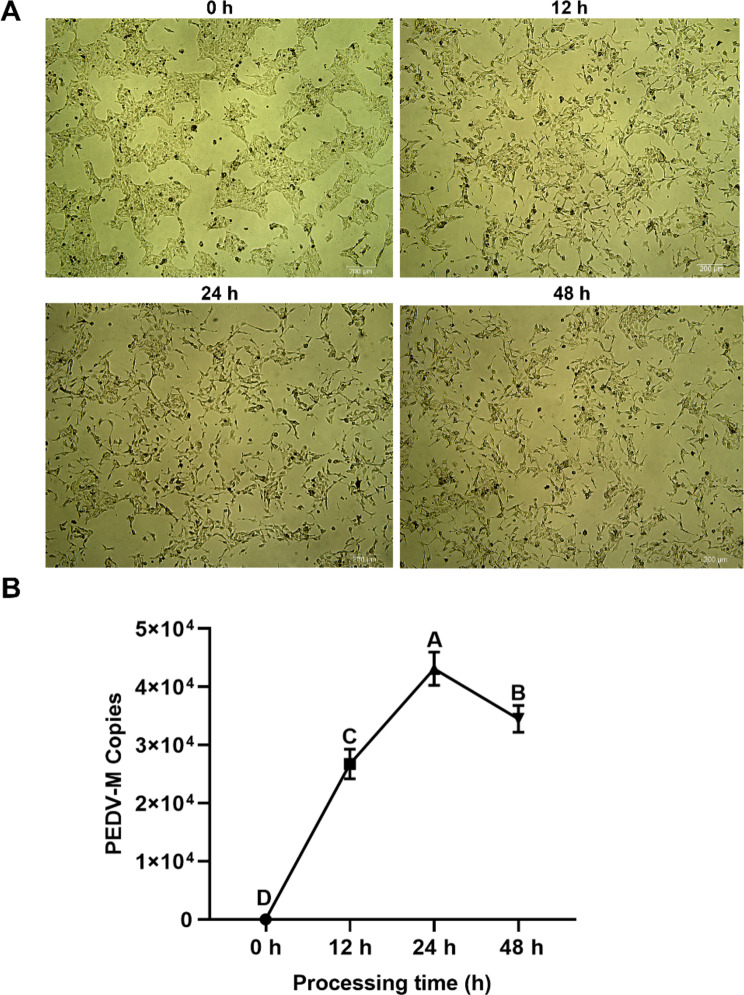
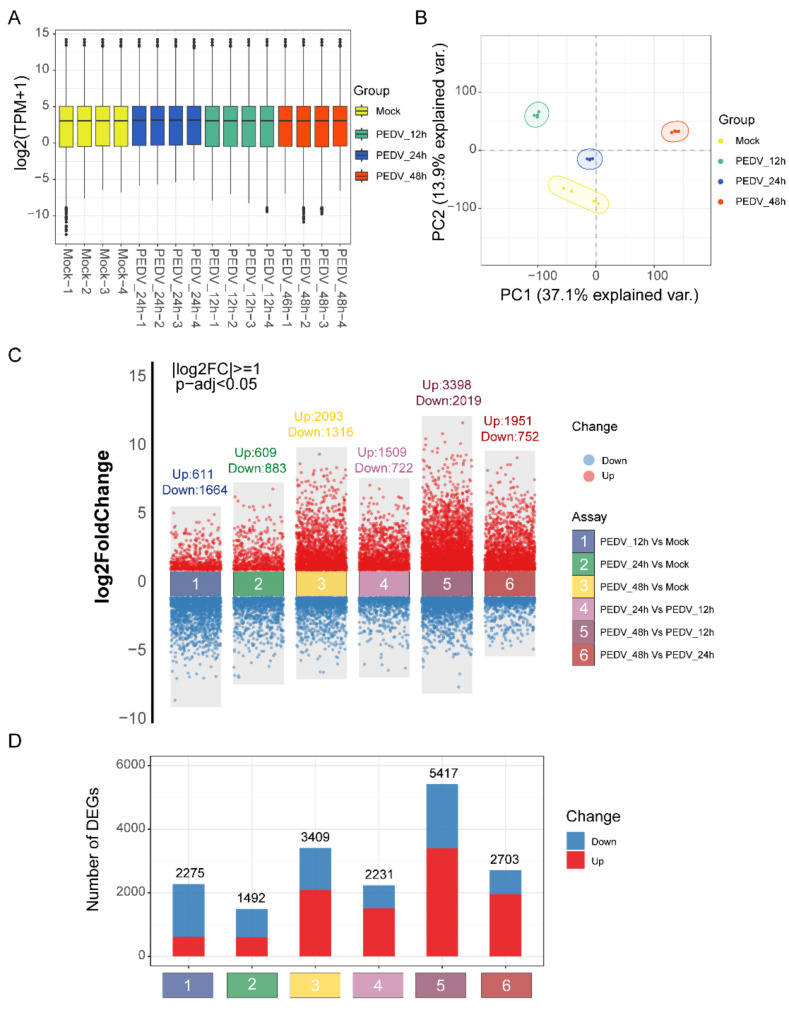
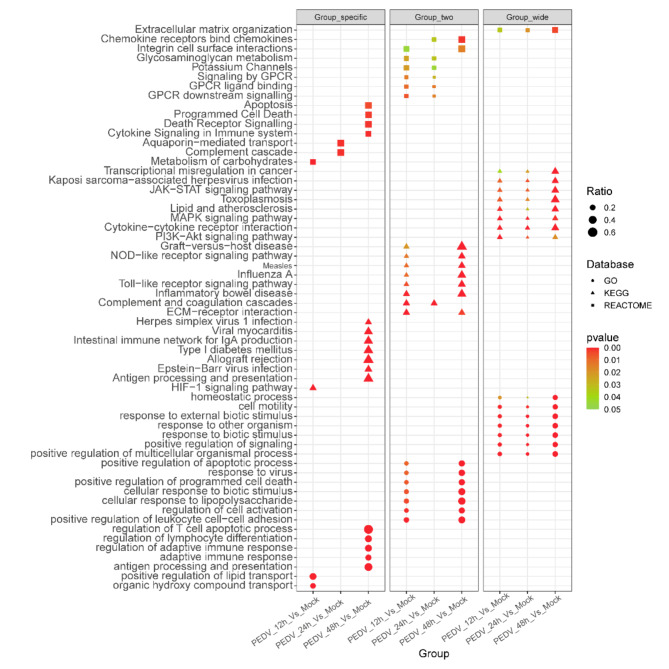
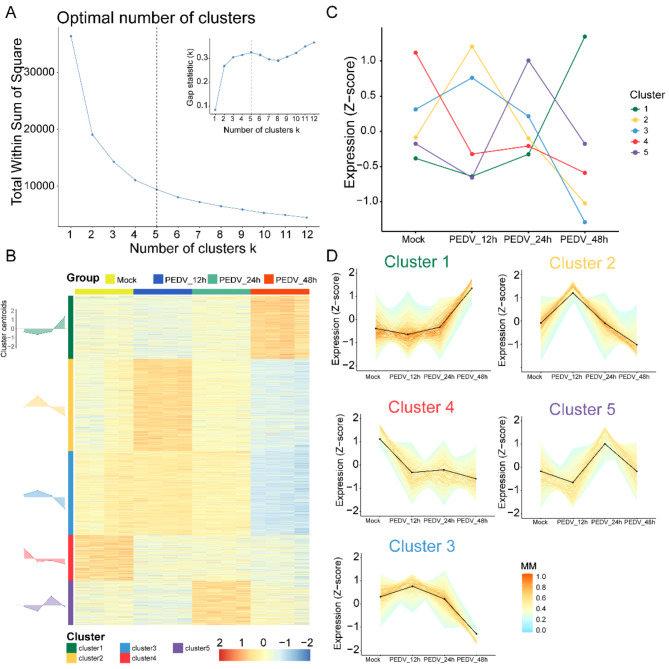
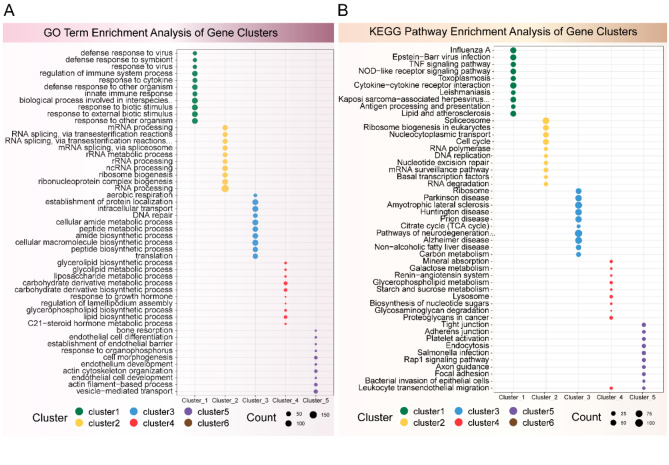
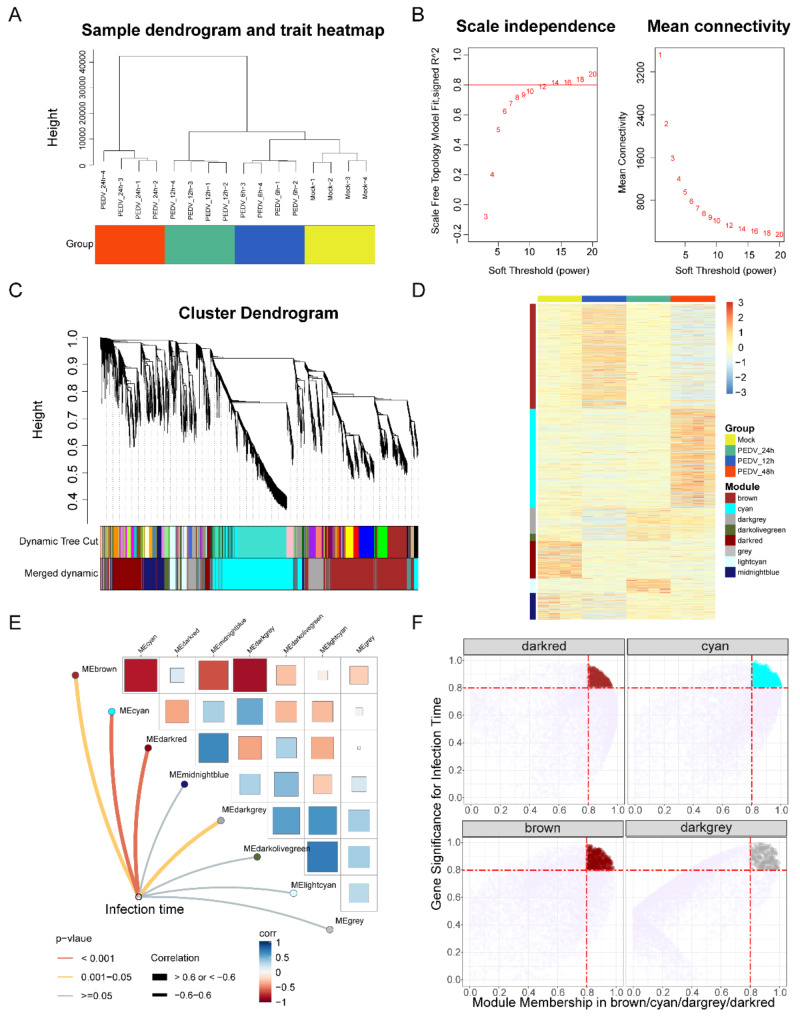
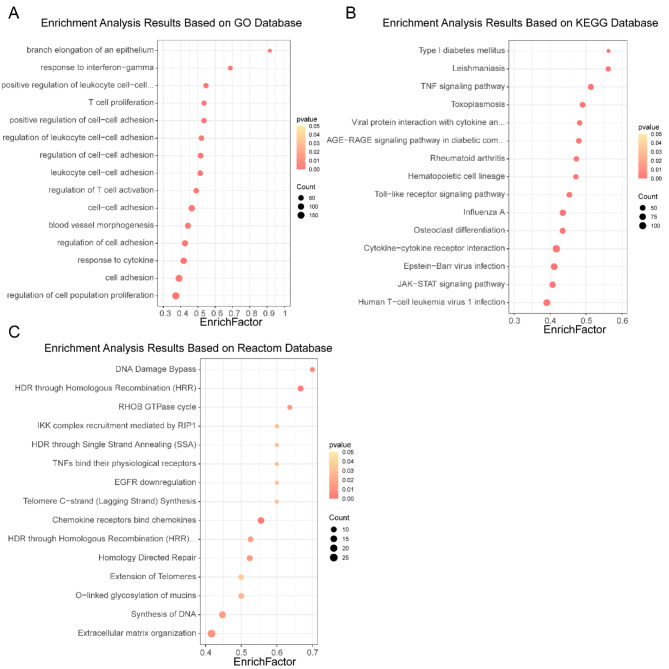
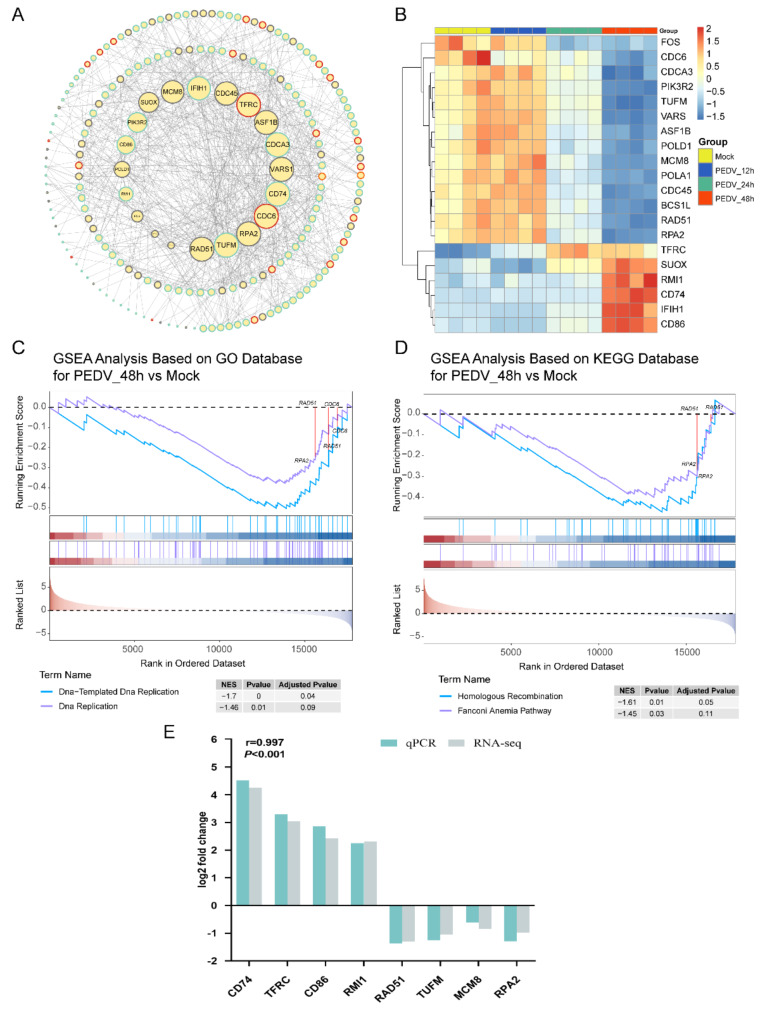
PEDV is a highly contagious enteric pathogen that can cause severe diarrhea and death in neonatal pigs. Despite extensive research, the molecular mechanisms of host's response to PEDV infection remain unclear. In this study, differentially expressed genes (DEGs), time-specific coexpression modules, and key regulatory genes associated with PEDV infection were identified. The analysis revealed 2,275, 1,492, and 3,409 DEGs in infected vs. mock-treated pigs at 12 h, 24 h, and 48 h, respectively. Time series analysis revealed that the upregulated genes were involved mainly in antiviral pathways such as the viral defense response and the regulation of immune system processes. Protein-protein interaction network analysis identified the top 20 core genes in the interaction network, which included six upregulated genes (TFRC, SUOX, RMI1, CD74, IFIH1, and CD86) and 14 downregulated genes (FOS, CDC6, CDCA3, PIK3R2, TUFM, VARS, ASF1B, POLD1, MCM8, POLA1, CDC45, BCS1L, RAD51, and RPA2). In addition, GSEA enrichment analysis revealed that pathways such as DNA replication and homologous recombination involving RAD51, CDC6, and RPA2 were significantly inhibited during viral infection. Our findings not only reveal dynamic changes in the transcriptome profile of PEDV-infected IPEC-J2 cells but also provide novel insights into the mechanism of PEDV infection of the host.
Keywords: DEGs; IPEC-J2; Molecular mechanism; PEDV; Transcriptome.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
-
- Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. 1997;142:629–33. - PubMed
-
- Cheun-Arom T, Temeeyasen G, Tripipat T, Kaewprommal P, Piriyapongsa J, Sukrong S, Chongcharoen W, Tantituvanont A, Nilubol D. Full-length genome analysis of two genetically distinct variants of porcine epidemic diarrhea virus in Thailand. Infect Genet Evol. 2016;44:114–21. 10.1016/j.meegid.2016.06.046. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 2021kj18/The Youth Support Project of Jiangsu Vocational College of Agriculture and Forestry
- 2021kj18/The Youth Support Project of Jiangsu Vocational College of Agriculture and Forestry
- 21KJB230003/The Basic Science (Natural Science) Research Project of Colleges in Jiangsu Province
- 21KJB230003/The Basic Science (Natural Science) Research Project of Colleges in Jiangsu Province
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
