

Ca2+ signaling in vascular smooth muscle and endothelial cells in blood vessel remodeling: a review
- PMID: 39731196
- PMCID: PMC11673324
- DOI: 10.1186/s41232-024-00363-0
Ca2+ signaling in vascular smooth muscle and endothelial cells in blood vessel remodeling: a review
Abstract
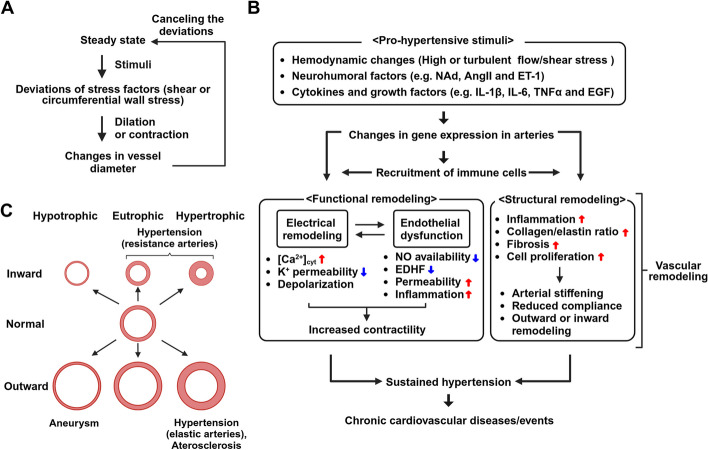
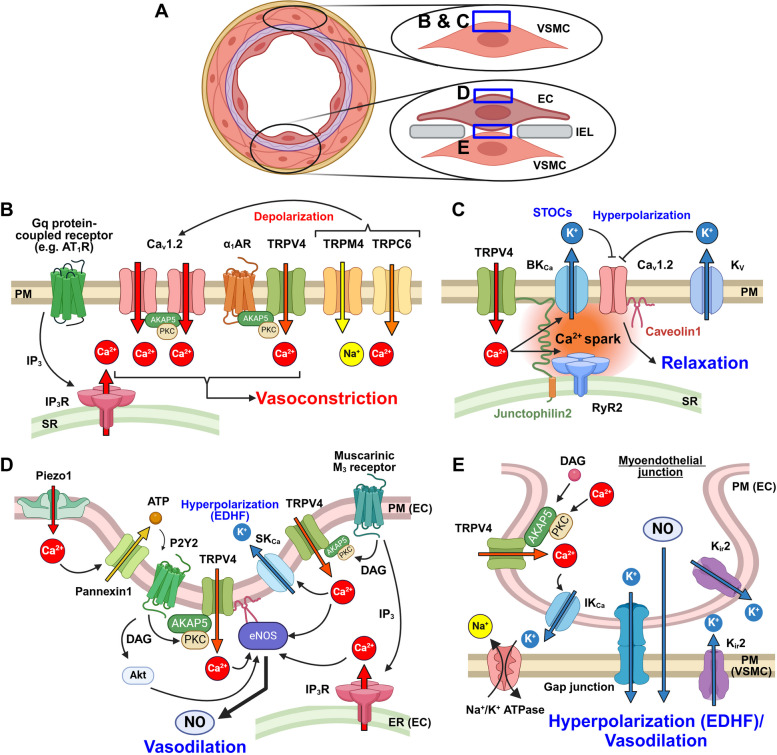
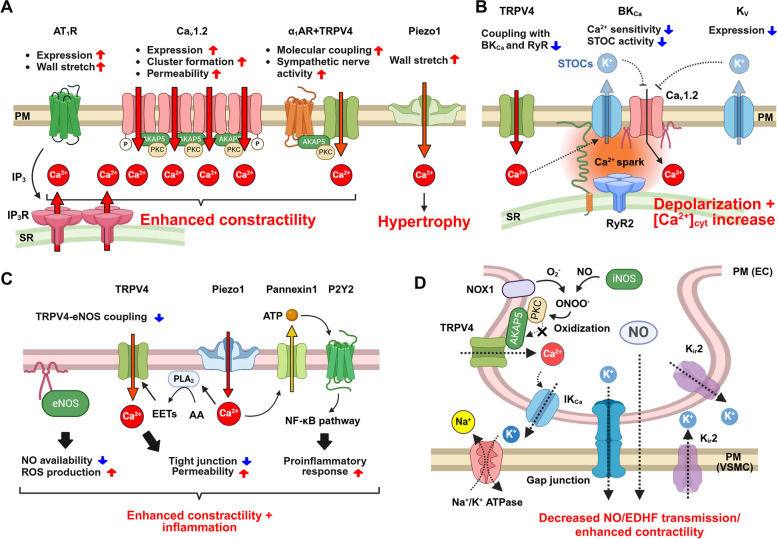
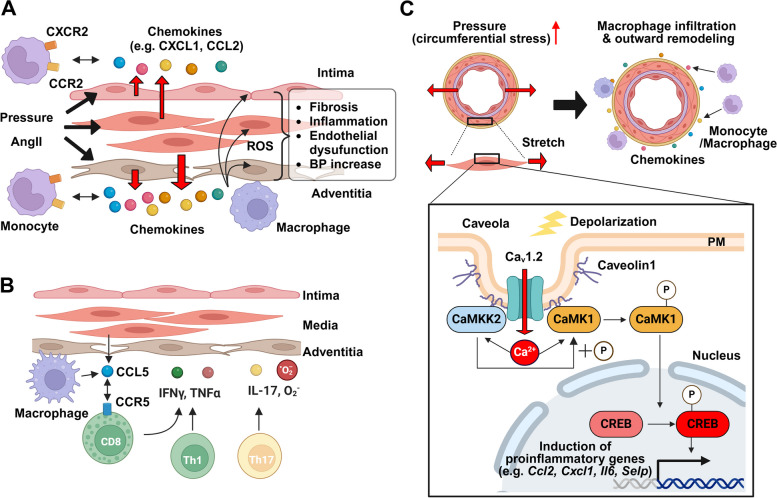
Vascular smooth muscle cells (VSMCs) and endothelial cells (ECs) act together to regulate blood pressure and systemic blood flow by appropriately adjusting blood vessel diameter in response to biochemical or biomechanical stimuli. Ion channels that are expressed in these cells regulate membrane potential and cytosolic Ca2+ concentration ([Ca2+]cyt) in response to such stimuli. The subsets of these ion channels involved in Ca2+ signaling often form molecular complexes with intracellular molecules via scaffolding proteins. This allows Ca2+ signaling to be tightly controlled in localized areas within the cell, resulting in a balanced vascular tone. When hypertensive stimuli are applied to blood vessels for extended periods, gene expression in these vascular cells can change dramatically. For example, alteration in ion channel expression often induces electrical remodeling that produces a depolarization of the membrane potential and elevated [Ca2+]cyt. Coupled with endothelial dysfunction blood vessels undergo functional remodeling characterized by enhanced vasoconstriction. In addition, pathological challenges to vascular cells can induce inflammatory gene products that may promote leukocyte infiltration, in part through Ca2+-dependent pathways. Macrophages accumulating in the vascular adventitia promote fibrosis through extracellular matrix turnover, and cause structural remodeling of blood vessels. This functional and structural remodeling often leads to chronic hypertension affecting not only blood vessels, but also multiple organs including the brain, kidneys, and heart, thus increasing the risk of severe cardiovascular events. In this review, we outline recent advances in multidisciplinary research concerning Ca2+ signaling in VSMCs and ECs, with an emphasis on the mechanisms underlying functional and structural vascular remodeling.
Keywords: Ca2+ signaling; Endothelial cells; Hypertension; Macrophages; Monocytes; Vascular remodeling; Vascular smooth muscle cells.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that they have no competing interests.
Figures
Similar articles
-
Ca2+ microdomains in vascular smooth muscle cells: Roles in vascular tone regulation and hypertension.J Pharmacol Sci. 2025 May;158(1):59-67. doi: 10.1016/j.jphs.2025.03.008. Epub 2025 Mar 14. J Pharmacol Sci. 2025. PMID: 40121058 Review.
-
Differences in TRPC3 and TRPC6 channels assembly in mesenteric vascular smooth muscle cells in essential hypertension.J Physiol. 2017 Mar 1;595(5):1497-1513. doi: 10.1113/JP273327. Epub 2016 Dec 29. J Physiol. 2017. PMID: 27861908 Free PMC article.
-
Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling.Compr Physiol. 2011 Jul;1(3):1555-602. doi: 10.1002/cphy.c100023. Compr Physiol. 2011. PMID: 23733654 Free PMC article. Review.
-
The role and mechanism of vascular wall cell ion channels in vascular fibrosis remodeling.Channels (Austin). 2024 Dec;18(1):2418128. doi: 10.1080/19336950.2024.2418128. Epub 2024 Oct 19. Channels (Austin). 2024. PMID: 39425532 Free PMC article. Review.
-
Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension.Am J Physiol Cell Physiol. 2014 Aug 15;307(4):C373-83. doi: 10.1152/ajpcell.00115.2014. Epub 2014 Jun 11. Am J Physiol Cell Physiol. 2014. PMID: 24920677 Free PMC article.
Cited by
-
Advances in magnetic field approaches for non-invasive targeting neuromodulation.Front Hum Neurosci. 2025 Apr 28;19:1489940. doi: 10.3389/fnhum.2025.1489940. eCollection 2025. Front Hum Neurosci. 2025. PMID: 40356879 Free PMC article. Review.
-
Comparative Analysis of ICS Combined with LABA versus Addition of Omalizumab on Transcriptomic Expression Profiles in Patients with Allergic Asthma.J Asthma Allergy. 2025 Jun 5;18:941-954. doi: 10.2147/JAA.S511885. eCollection 2025. J Asthma Allergy. 2025. PMID: 40491983 Free PMC article.
-
Ca2+ microdomain-based excitation-transcription coupling in cardiac myocytes and vascular smooth muscle cells.Inflamm Regen. 2025 Jun 23;45(1):19. doi: 10.1186/s41232-025-00384-3. Inflamm Regen. 2025. PMID: 40551210 Free PMC article. Review.
-
The Calcium Signalling Profile of the Inner Blood-Retinal Barrier in Diabetic Retinopathy.Cells. 2025 Jun 6;14(12):856. doi: 10.3390/cells14120856. Cells. 2025. PMID: 40558483 Free PMC article. Review.
References
-
- Lacolley P, Regnault V, Segers P, Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97(4):1555–617. 10.1152/physrev.00003.2017. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
