

Inactivation of the SLC25A1 gene during embryogenesis induces a unique senescence program controlled by p53
- PMID: 39733217
- PMCID: PMC12089371
- DOI: 10.1038/s41418-024-01428-w
Inactivation of the SLC25A1 gene during embryogenesis induces a unique senescence program controlled by p53
Abstract
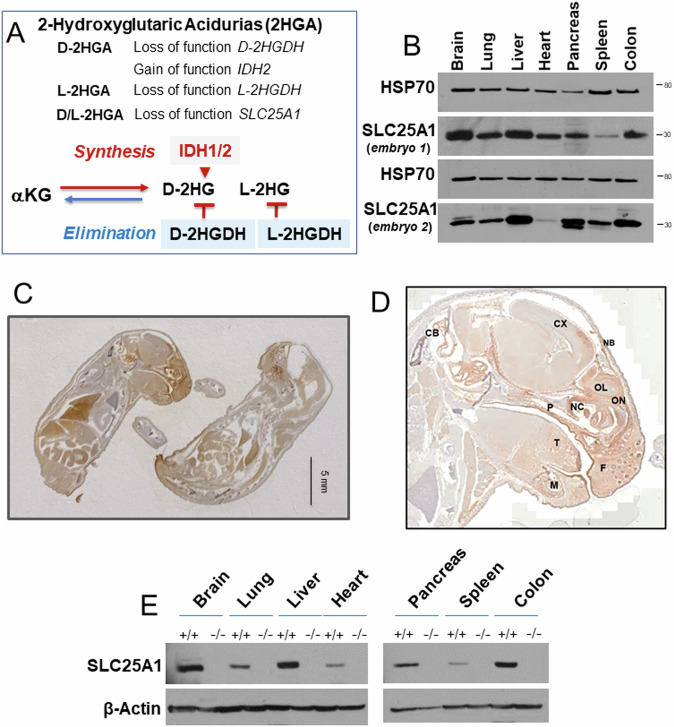
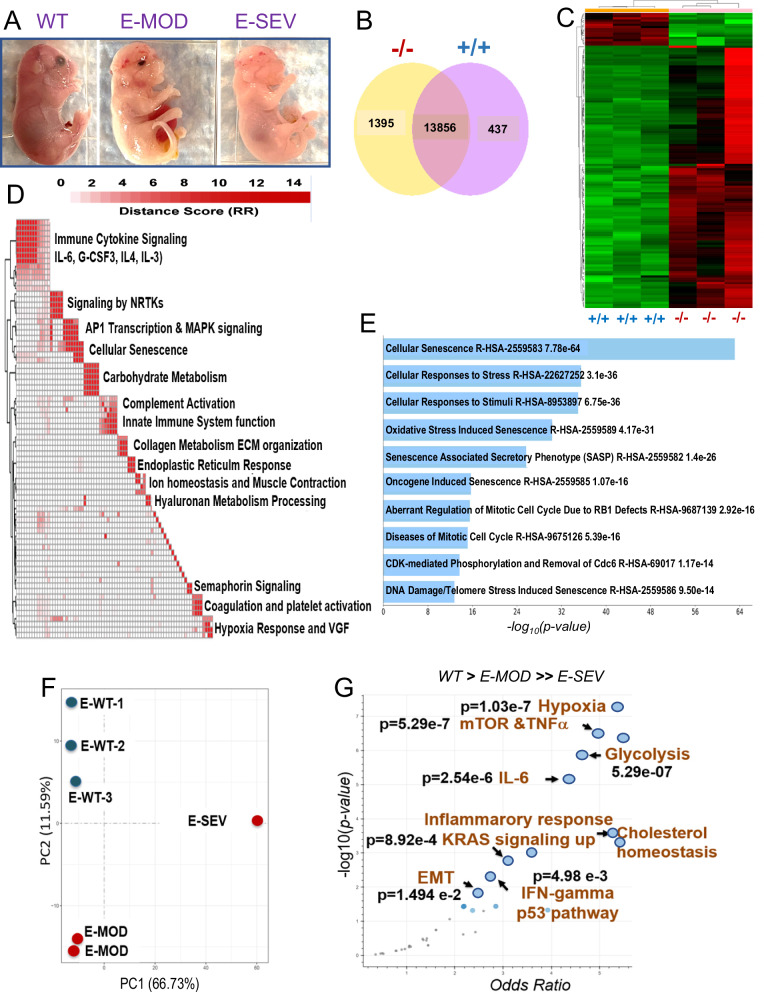
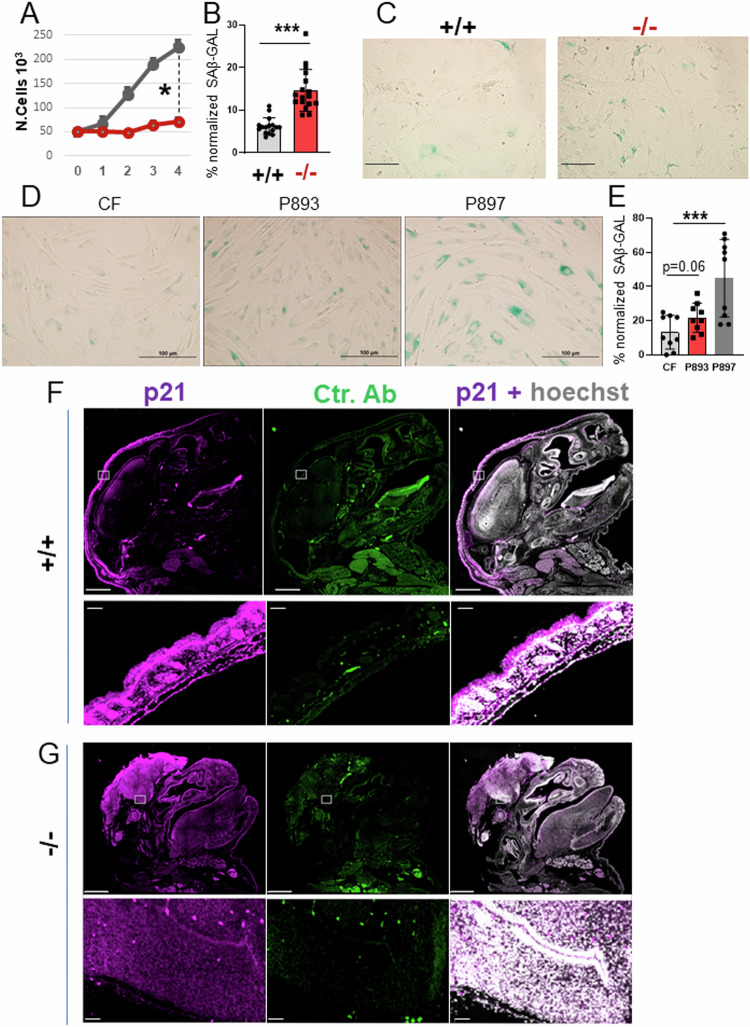
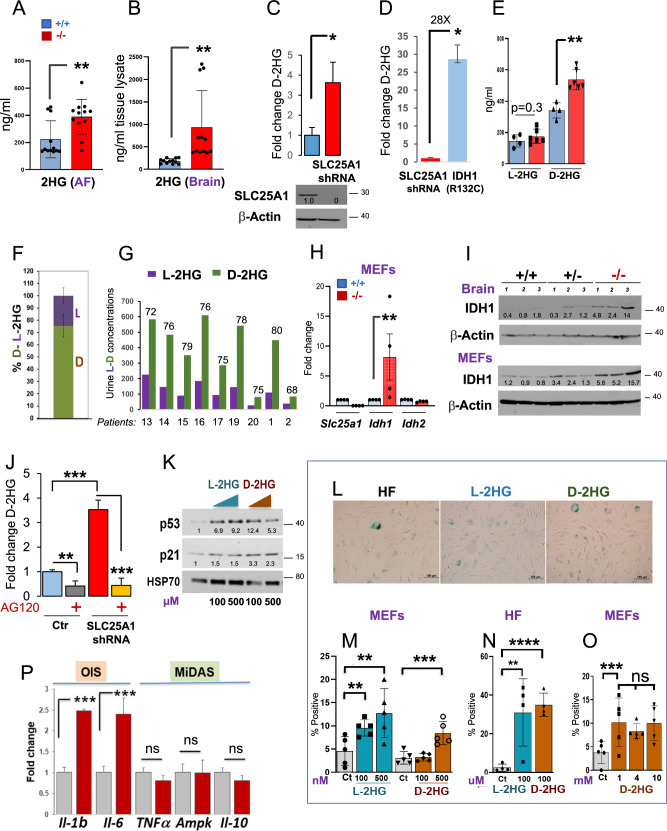
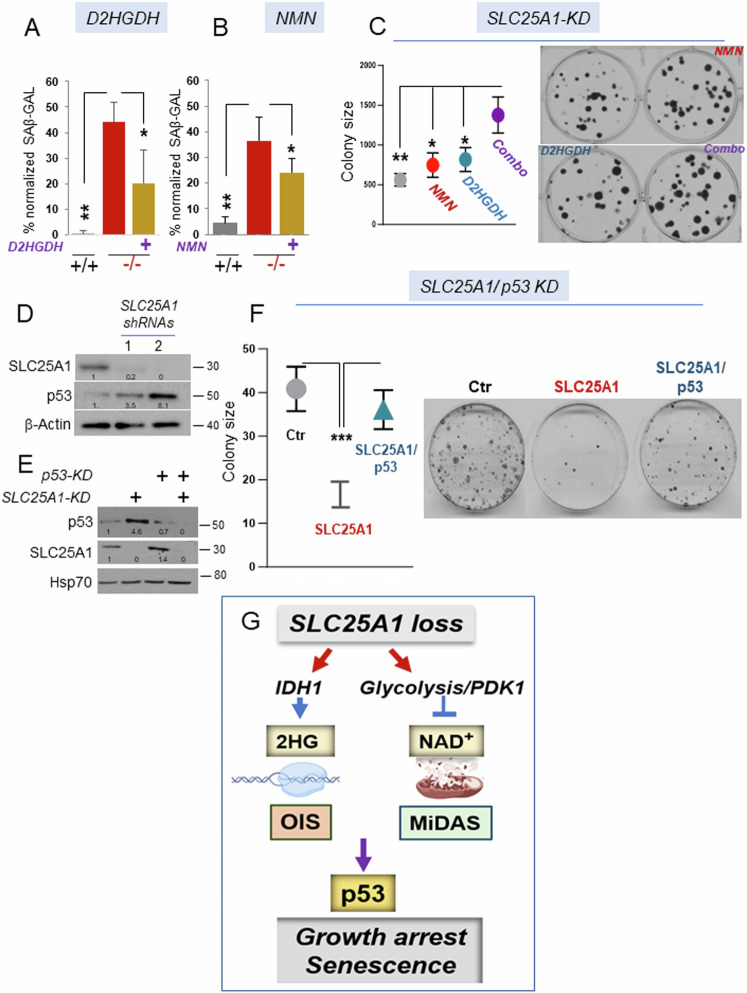
Germline inactivating mutations of the SLC25A1 gene contribute to various human disorders, including Velocardiofacial (VCFS), DiGeorge (DGS) syndromes and combined D/L-2-hydroxyglutaric aciduria (D/L-2HGA), a severe systemic disease characterized by the accumulation of 2-hydroxyglutaric acid (2HG). The mechanisms by which SLC25A1 loss leads to these syndromes remain largely unclear. Here, we describe a mouse model of SLC25A1 deficiency that mimics human VCFS/DGS and D/L-2HGA. Surprisingly, inactivation of both Slc25a1 alleles results in alterations in the development of multiple organs, and in a severe proliferation defect by activating two senescence programs, oncogene-induced senescence (OIS) and mitochondrial dysfunction-induced senescence (MiDAS), which converge upon the induction of the p53 tumor suppressor. Mechanistically, cells and tissues with dysfunctional SLC25A1 protein undergo metabolic and transcriptional rewiring leading to the accumulation of 2HG via a non-canonical pathway and to the depletion of nicotinamide adenine dinucleotide, NAD+, which trigger senescence. Replenishing the pool of NAD+ or promoting the clearance of 2HG rescues the proliferation defect of cells with dysfunctional SLC25A1 in a cooperative fashion. Further, removal of p53 activity via RNA interference restores proliferation, indicating that p53 acts as a critical barrier to the expansion of cells lacking functional SLC25A1. These findings reveal unexpected pathogenic roles of senescence and of p53 in D/L-2HGA and identify potential therapeutic strategies to correct salient molecular alterations driving this disease.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethical approval and consent to participate: All animal studies were conducted in compliance with ethical regulations according to protocol #2017-1192 approved by the Institutional Animal Care and Use Committee (IACUC) at Georgetown University. All the mice were housed at Georgetown University Division of Comparative Medicine, in a SPF vivarium which is maintained at a 12:12 h light:dark cycle, at 68–74 °F and 30–70% humidity range, according to IACUC regulations. Mice were euthanized according to the IACUC guidelines. This study does not directly involve human subjects or human data that requires ethical approval. The collection and use of atient-derived samples utilized herein was approved by the Institutional Review Board at the University of Pittsburgh, protocol #19090211 [34].
Figures





Update of
-
Loss of the mitochondrial carrier, SLC25A1, during embryogenesis induces a unique senescence program controlled by p53.bioRxiv [Preprint]. 2024 May 31:2023.07.18.549409. doi: 10.1101/2023.07.18.549409. bioRxiv. 2024. Update in: Cell Death Differ. 2025 May;32(5):818-836. doi: 10.1038/s41418-024-01428-w. PMID: 37503155 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
