

The combination of decitabine with multi-omics confirms the regulatory pattern of the correlation between DNA methylation of the CACNA1C gene and atrial fibrillation
- PMID: 39734414
- PMCID: PMC11681619
- DOI: 10.3389/fphar.2024.1497977
The combination of decitabine with multi-omics confirms the regulatory pattern of the correlation between DNA methylation of the CACNA1C gene and atrial fibrillation
Abstract
Background: Studies have shown that DNA methylation of the CACNA1C gene is involved in the pathogenesis of various diseases and the mechanism of drug action. However, its relationship with atrial fibrillation (AF) remains largely unexplored.
Objective: To investigate the association between DNA methylation of the CACNA1C gene and AF by combining decitabine (5-Aza-2'-deoxycytidine, AZA) treatment with multi-omics analysis.
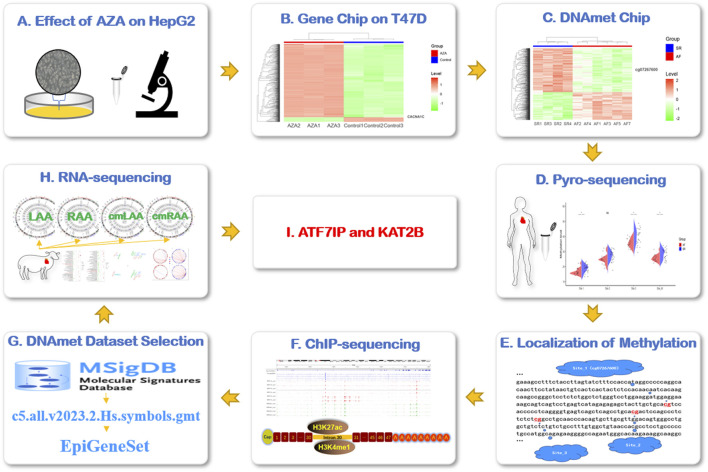
Methods: HepG2 cells were treated with AZA to observe the expression of the CACNA1C gene, which was further validated using gene expression microarrays. Pyrosequencing was employed to validate differentially methylated sites of the CACNA1C gene observed in DNA methylation microarrays. A custom DNA methylation dataset based on the MSigDB database was combined with ChIP-sequencing and RNA-sequencing data to explore the regulatory patterns of DNA methylation of the CACNA1C gene.
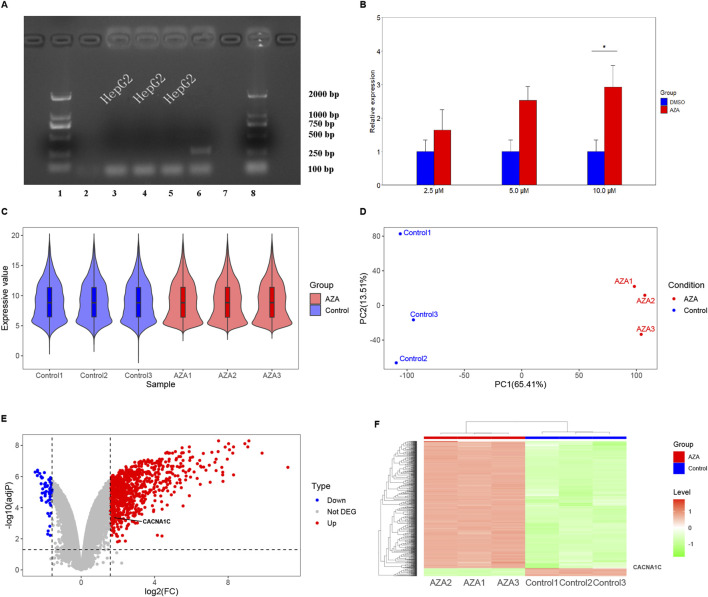
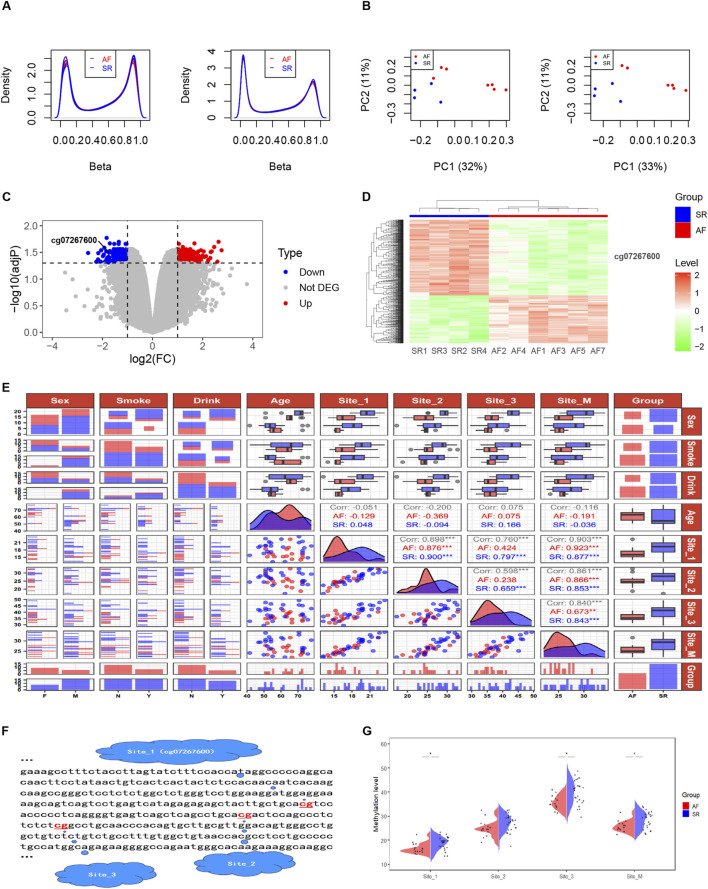
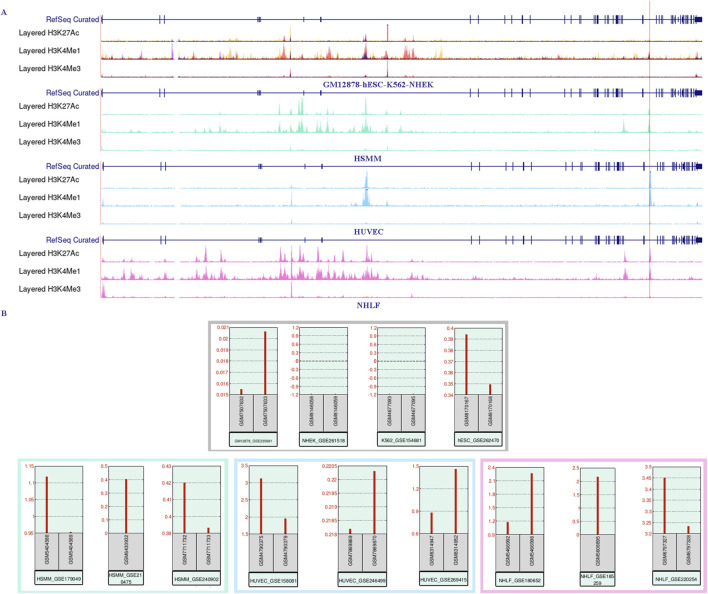
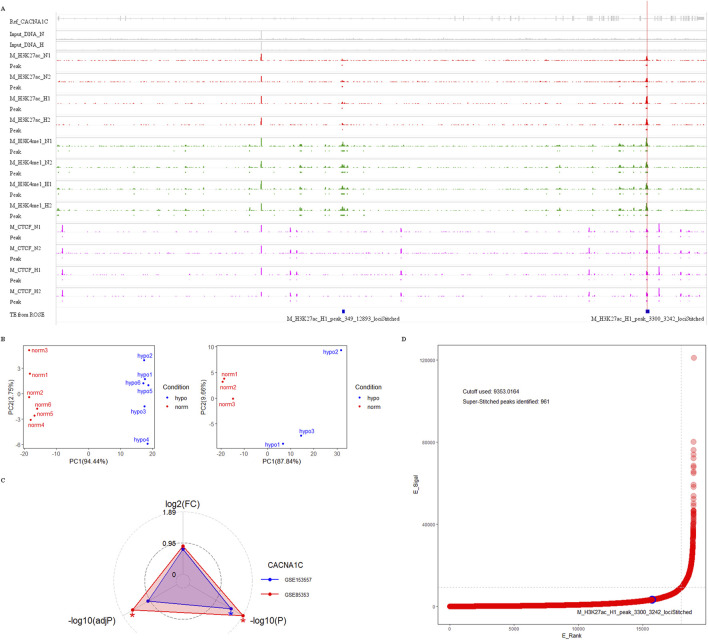
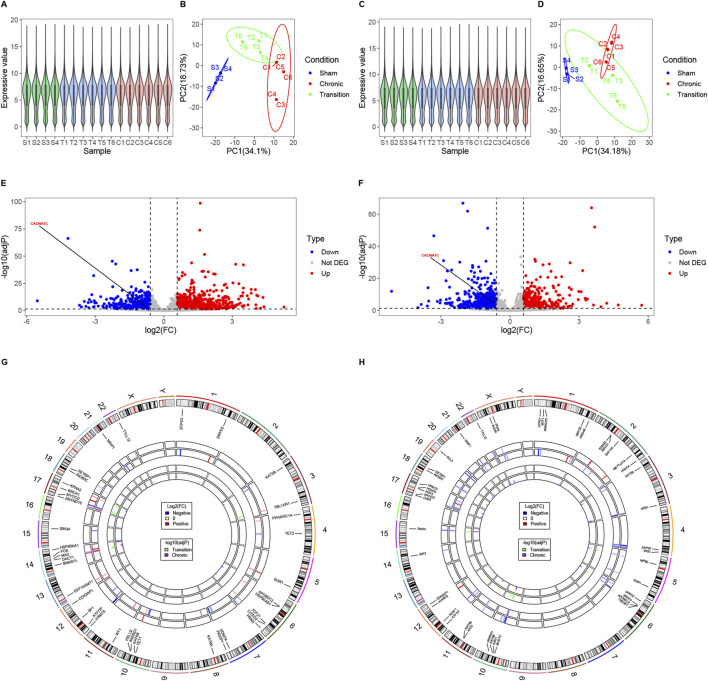
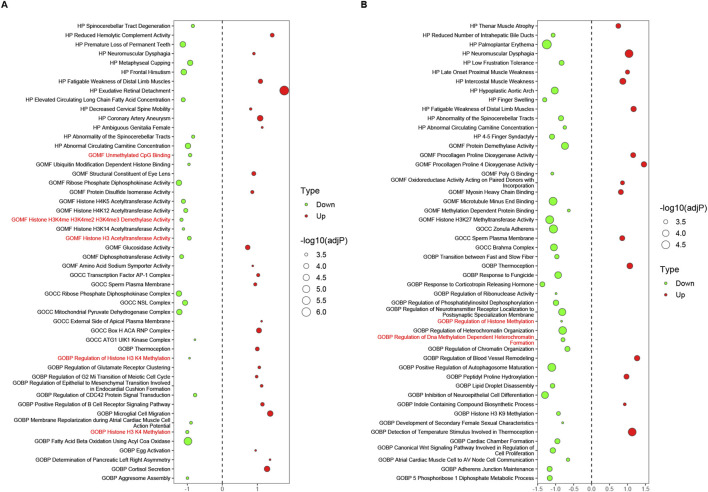
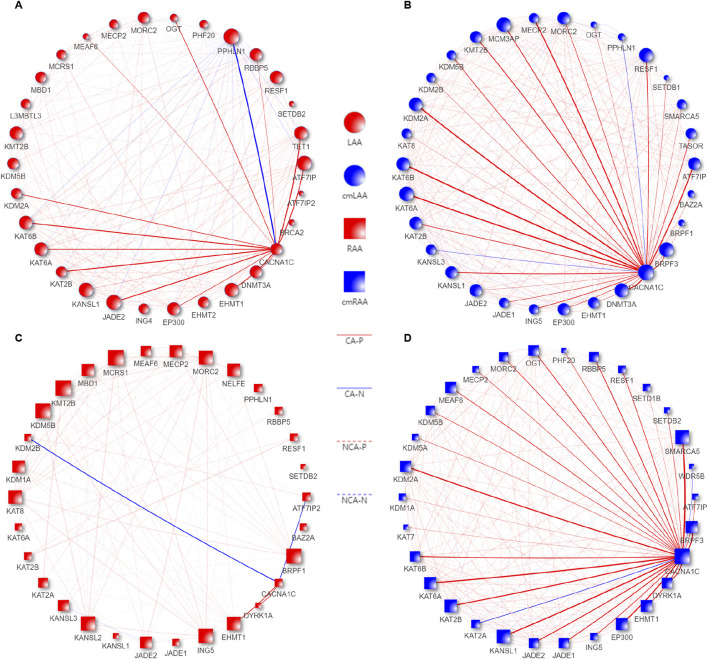
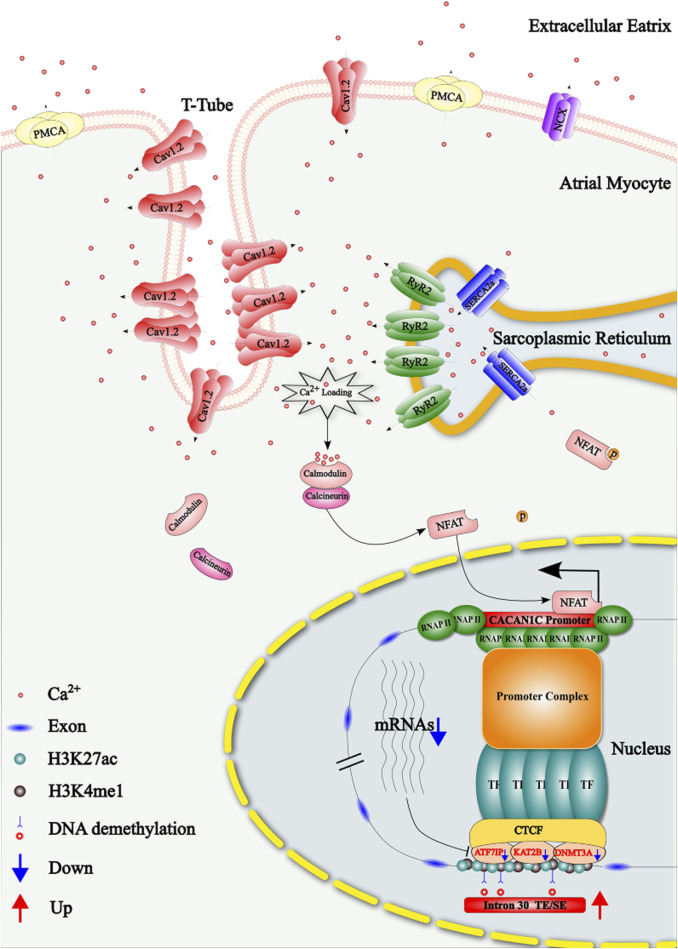
Results: Treatment of HepG2 cells with three different concentrations of AZA (2.5 µM, 5.0 µM, and 10.0 µM) resulted in 1.6, 2.5, and 2.9-fold increases in the mRNA expression of the CACNA1C gene, respectively, compared to the DMSO group, with statistical significance at the highest concentration group (p < 0.05). Similarly, AZA treatment of T47D cells showed upregulated mRNA expression of the CACNA1C gene in the gene expression microarray results (adj P < 0.05). DNA methylation microarray analysis revealed that methylation of a CpG site in intron 30 of the CACNA1C gene may be associated with AF (adj P < 0.05). Pyrosequencing of this site and its adjacent two CpG sites demonstrated significant differences in DNA methylation levels between AF and sinus rhythm groups (p < 0.05). Subsequent multivariate logistic regression models confirmed that the DNA methylation degree of these three sites and their average was associated with AF (p < 0.05). Additionally, the UCSC browser combined with ChIP-sequencing revealed that the aforementioned region was enriched in enhancer markers H3K27ac and H3K4me1. Differential expression and pathway analysis of RNA-sequencing data ultimately identified ATF7IP and KAT2B genes as potential regulators of the CACNA1C gene.
Conclusion: The DNA methylation levels at three CpG sites in intron 30 of the CACNA1C gene are associated with AF status, and potentially regulated by ATF7IP and KAT2B.
Keywords: DNA methylation; Pyro-sequencing; atrial fibrillation; calcium voltage-gated channel subunit alpha1 C; decitabine.
Copyright © 2024 Yang, Li, Liu, Shao, Yang, Niu, Peng and Meng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Genome-wide DNA methylation analysis in permanent atrial fibrillation.Mol Med Rep. 2017 Oct;16(4):5505-5514. doi: 10.3892/mmr.2017.7221. Epub 2017 Aug 10. Mol Med Rep. 2017. PMID: 28849195
-
Demethylation by 5-aza-2'-deoxycytidine in colorectal cancer cells targets genomic DNA whilst promoter CpG island methylation persists.BMC Cancer. 2010 Jul 12;10:366. doi: 10.1186/1471-2407-10-366. BMC Cancer. 2010. PMID: 20618997 Free PMC article.
-
[mRNA genomics change and significance of important ion channel proteins in patients with atrial fibrillation].Zhonghua Yi Xue Za Zhi. 2018 Oct 23;98(39):3171-3177. doi: 10.3760/cma.j.issn.0376-2491.2018.39.009. Zhonghua Yi Xue Za Zhi. 2018. PMID: 30392277 Chinese.
-
Global demethylation of rat chondrosarcoma cells after treatment with 5-aza-2'-deoxycytidine results in increased tumorigenicity.PLoS One. 2009 Dec 17;4(12):e8340. doi: 10.1371/journal.pone.0008340. PLoS One. 2009. PMID: 20019818 Free PMC article.
-
Genome-wide methylation and expression profiling identifies promoter characteristics affecting demethylation-induced gene up-regulation in melanoma.BMC Med Genomics. 2010 Feb 9;3:4. doi: 10.1186/1755-8794-3-4. BMC Med Genomics. 2010. PMID: 20144234 Free PMC article.
References
LinkOut - more resources
Full Text Sources