

MSA clustering enhances AF-Multimer's ability to predict conformational landscapes of protein-protein interactions
- PMID: 39735576
- PMCID: PMC11671036
- DOI: 10.1093/bioadv/vbae197
MSA clustering enhances AF-Multimer's ability to predict conformational landscapes of protein-protein interactions
Abstract
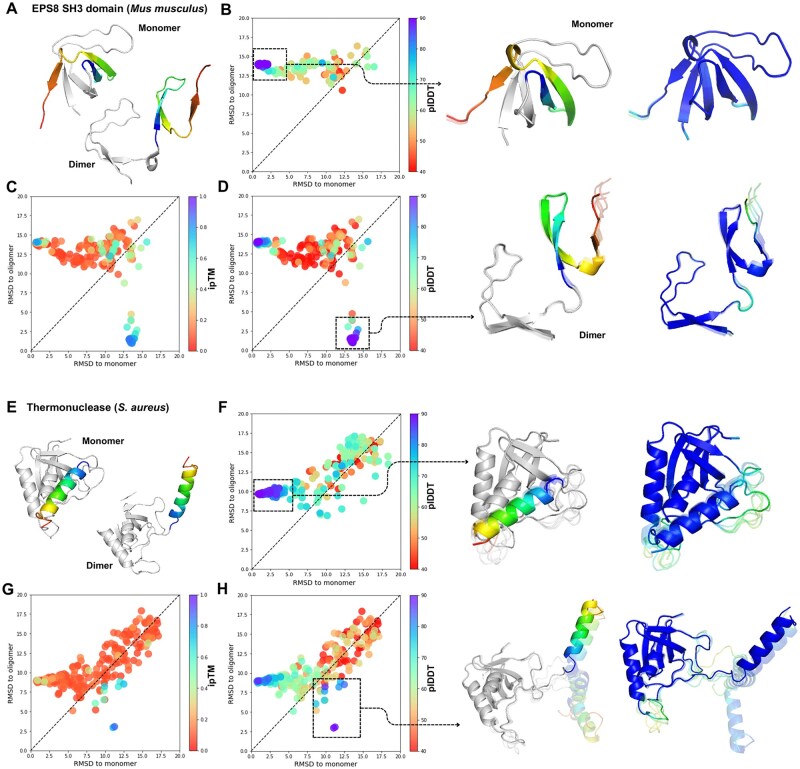
Motivation: Understanding the conformational landscape of protein-ligand interactions is critical for elucidating the binding mechanisms that govern these interactions. Traditional methods like molecular dynamics (MD) simulations are computationally intensive, leading to a demand for more efficient approaches. This study explores how multiple sequence alignment (MSA) clustering enhance AF-Multimer's ability to predict conformational landscapes, particularly for proteins with multiple conformational states.
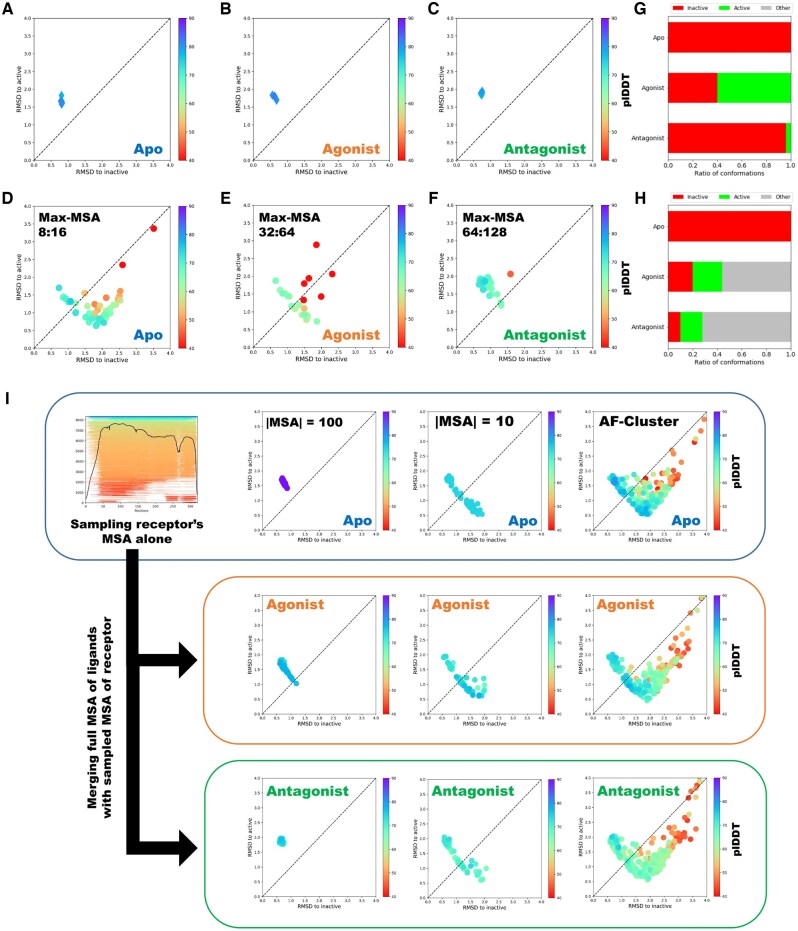
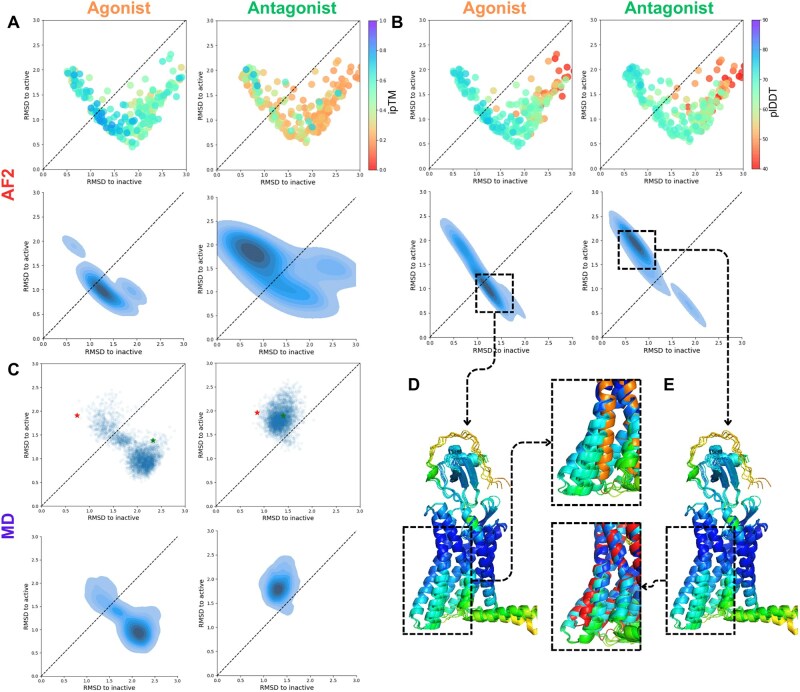
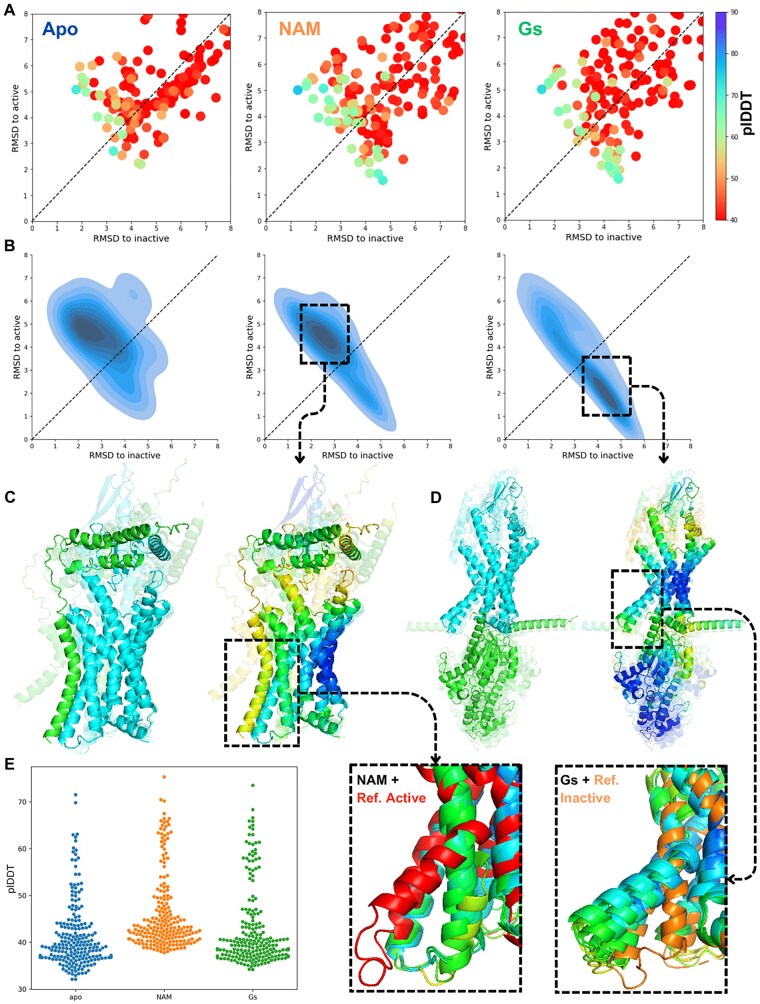
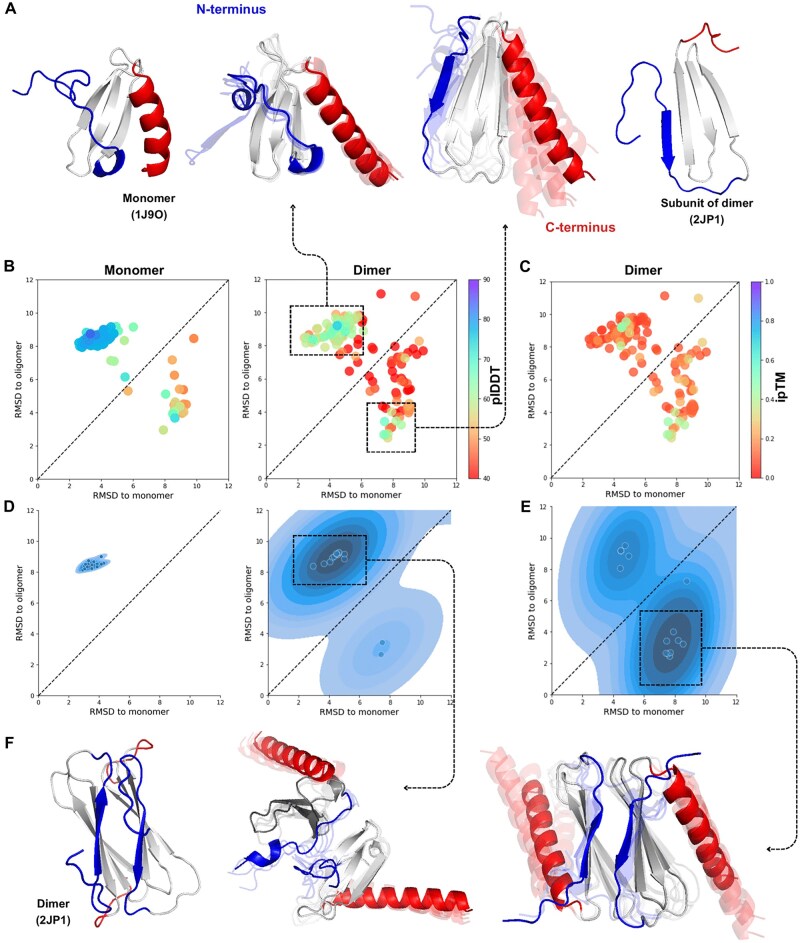
Results: We verified this approach by predicting the conformational landscapes of chemokine receptor 4 (CXCR4) and glucagon receptor (GCGR) in the presence of their agonists and antagonists. In our experiments, AF-Multimer predicted the structures of CXCR4 and GCGR predominantly in active state in the presence of agonists and in inactive state in the presence of antagonists. Moreover, we tested our approach with proteins known to switch between monomeric and dimeric states, such as lymphotactin, SH3, and thermonuclease. AFcluster-Multimer accurately predicted conformational states during oligomerization, which AFcluster with AlphaFold2 alone fails to achieve. In conclusion, MSA clustering enhances AF-Multimer's ability to predict protein conformational landscapes and mechanistic effects of ligand binding, offering a robust tool for understanding protein-ligand interactions.
Availability and implementation: Code for running AFcluster-Multimer is available at https://github.com/KhondamirRustamov/AF-Multimer-cluster.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
References
-
- Abraham MJ, Murtola T, Schulz R et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015;1-2:19–25.
-
- Evans R, O’Neill M, Pritzel A . et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv, 2022, 10.1101/2021.10.04.463034. preprint: not peer reviewed. - DOI
LinkOut - more resources
Full Text Sources
