

Advancing Regulatory Genomics With Machine Learning
- PMID: 39735654
- PMCID: PMC11672376
- DOI: 10.1177/11779322241249562
Advancing Regulatory Genomics With Machine Learning
Abstract
In recent years, several machine learning (ML) approaches have been proposed to predict gene expression signal and chromatin features from the DNA sequence alone. These models are often used to deduce and, to some extent, assess putative new biological insights about gene regulation, and they have led to very interesting advances in regulatory genomics. This article reviews a selection of these methods, ranging from linear models to random forests, kernel methods, and more advanced deep learning models. Specifically, we detail the different techniques and strategies that can be used to extract new gene-regulation hypotheses from these models. Furthermore, because these putative insights need to be validated with wet-lab experiments, we emphasize that it is important to have a measure of confidence associated with the extracted hypotheses. We review the procedures that have been proposed to measure this confidence for the different types of ML models, and we discuss the fact that they do not provide the same kind of information.
Keywords: Regulatory genomics; deep learning; gene expression; machine learning; model interpretation; transcription factor binding sites.
© The Author(s) 2024.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
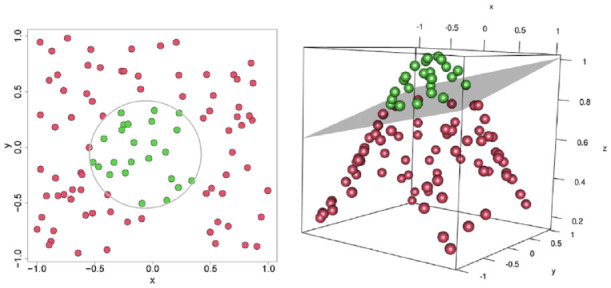
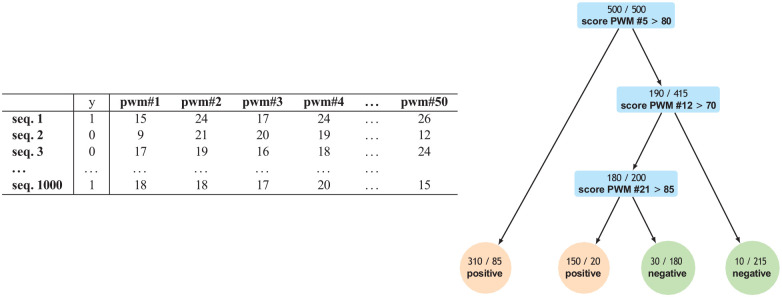


References
-
- Haussler D, Krogh A, Mian S, Sjolander K. Protein modeling using hidden Markov models: analysis of globins. Department of Computer and Information Sciences, University of California at Santa Cruz; 1992. Technical Report UCSC-CRL-92-23.
-
- Baldi P, Brunak S. Bioinformatics: The Machine Learning Approach. MIT Press; 1998.
-
- Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415-431. - PubMed
-
- Stormo GD. Consensus patterns in DNA. Meth Enzymol. 1990;183:211-221. - PubMed
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28-36. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
