

STLBRF: an improved random forest algorithm based on standardized-threshold for feature screening of gene expression data
- PMID: 39736135
- PMCID: PMC11735748
- DOI: 10.1093/bfgp/elae048
STLBRF: an improved random forest algorithm based on standardized-threshold for feature screening of gene expression data
Abstract
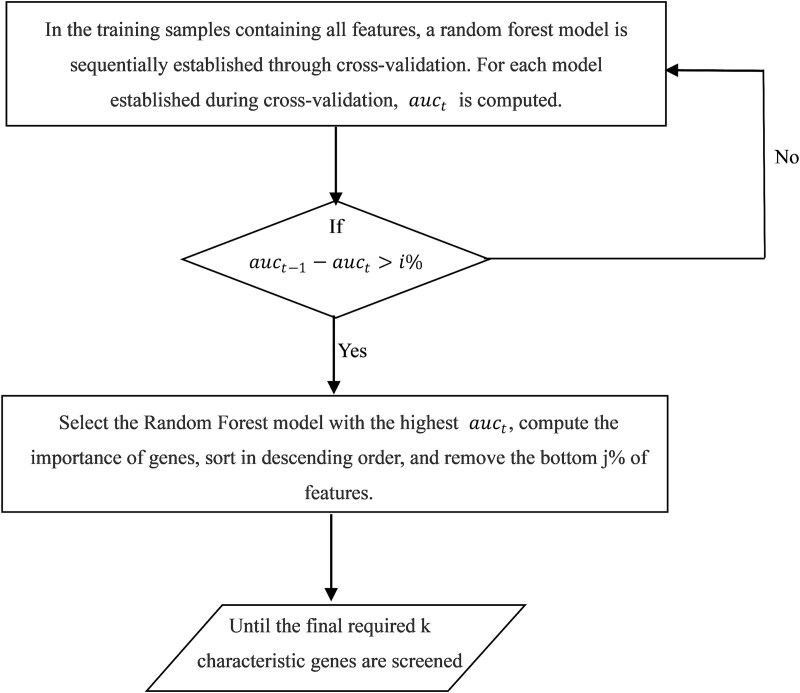
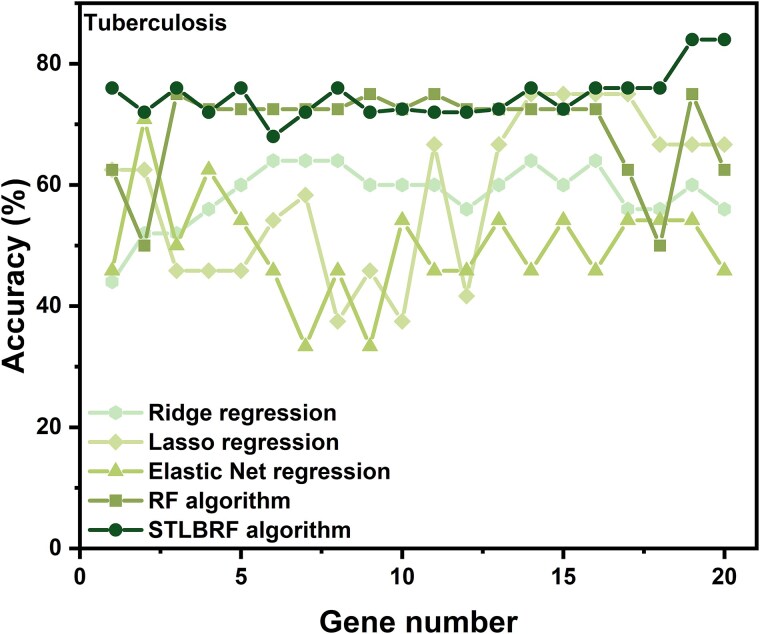
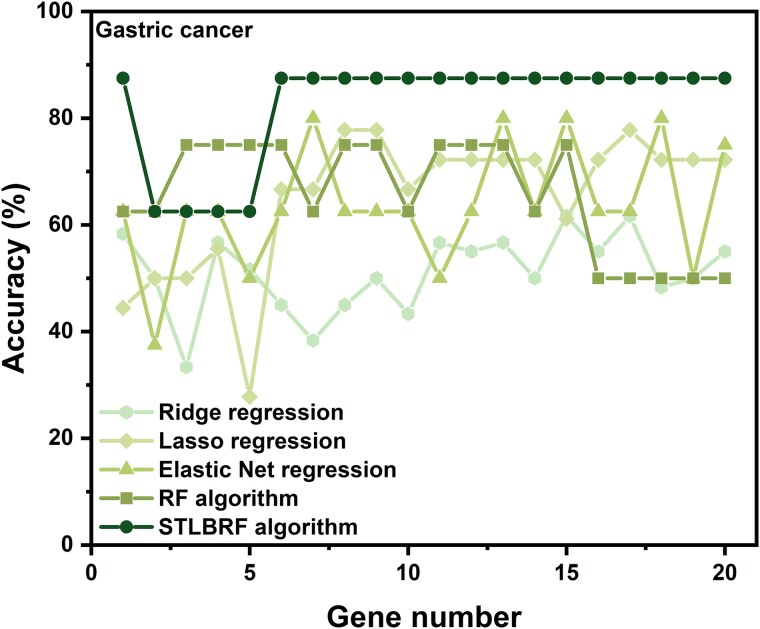
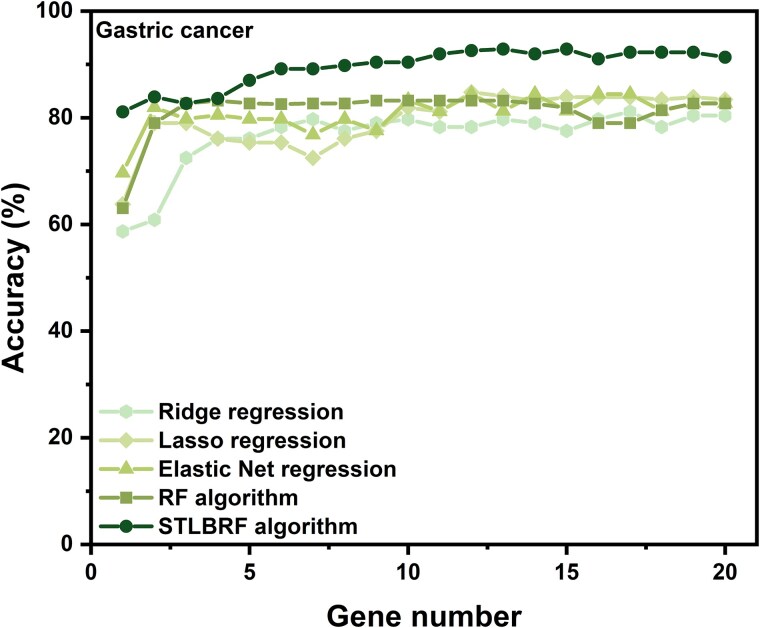
When the traditional random forest (RF) algorithm is used to select feature elements in biostatistical data, a large amount of noise data and parameters can affect the importance of the selected feature elements, making the control of feature selection difficult. Therefore, it is a challenge for the traditional RF algorithm to preserve the accuracy of algorithm results in the presence of noise data. Generally, directly removing noise data can result in significant bias in the results. In this study, we develop a new algorithm, standardized threshold, and loops based random forest (STLBRF), and apply it to the field of gene expression data for feature gene selection. This algorithm, based on the traditional RF algorithm, combines backward elimination and K-fold cross-validation to construct a cyclic system and set a standardized threshold: error increment. The algorithm overcomes the shortcomings of existing gene selection methods. We compare ridge regression, lasso regression, elastic net regression, the traditional RF algorithm, and our improved RF algorithm using three real gene expression datasets and conducting a quantitative analysis. To ensure the reliability of the results, we validate the effectiveness of the genes selected by these methods using the Random Forest classifier. The results indicate that, compared to other methods, the STLBRF algorithm achieves not only higher effectiveness in feature gene selection but also better control over the number of selected genes. Our method offers reliable technical support for feature expression analysis and research on biomarker selection.
Keywords: biomarker; feature gene selection; improved random forest algorithm; noise data; standardized threshold.
© The Author(s) 2024. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
A population spatialization method based on the integration of feature selection and an improved random forest model.PLoS One. 2025 Apr 3;20(4):e0321263. doi: 10.1371/journal.pone.0321263. eCollection 2025. PLoS One. 2025. PMID: 40179342 Free PMC article.
-
An Efficient Feature Selection Strategy Based on Multiple Support Vector Machine Technology with Gene Expression Data.Biomed Res Int. 2018 Aug 30;2018:7538204. doi: 10.1155/2018/7538204. eCollection 2018. Biomed Res Int. 2018. PMID: 30228989 Free PMC article.
-
Two-stage feature selection for classification of gene expression data based on an improved Salp Swarm Algorithm.Math Biosci Eng. 2022 Sep 19;19(12):13747-13781. doi: 10.3934/mbe.2022641. Math Biosci Eng. 2022. PMID: 36654066
-
G-Forest: An ensemble method for cost-sensitive feature selection in gene expression microarrays.Artif Intell Med. 2020 Aug;108:101941. doi: 10.1016/j.artmed.2020.101941. Epub 2020 Aug 14. Artif Intell Med. 2020. PMID: 32972668
-
Graph Random Forest: A Graph Embedded Algorithm for Identifying Highly Connected Important Features.Biomolecules. 2023 Jul 20;13(7):1153. doi: 10.3390/biom13071153. Biomolecules. 2023. PMID: 37509188 Free PMC article.
Cited by
-
Deciphering the Regulatory Networks of the Migrasome-Associated Cell Subpopulation in Heterotopic Ossification via Multi-Omics Analysis.FASEB J. 2025 Jun 30;39(12):e70749. doi: 10.1096/fj.202500965R. FASEB J. 2025. PMID: 40540299 Free PMC article.
References
-
- Nelson PT, Baldwin DA, Scearce LM. et al. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat Methods 2004;1:155–61. - PubMed
-
- Su YS, Li YX, Zhang Z. et al. Features identification for phenotypic classification based on genes and gene pairs. Curr Bioinform 2018;13:468–78.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
