

Descart: a method for detecting spatial chromatin accessibility patterns with inter-cellular correlations
- PMID: 39736655
- PMCID: PMC11686967
- DOI: 10.1186/s13059-024-03458-6
Descart: a method for detecting spatial chromatin accessibility patterns with inter-cellular correlations
Abstract
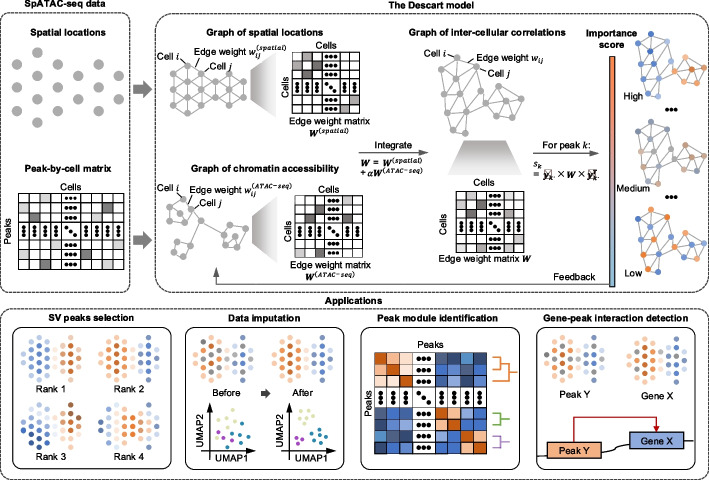
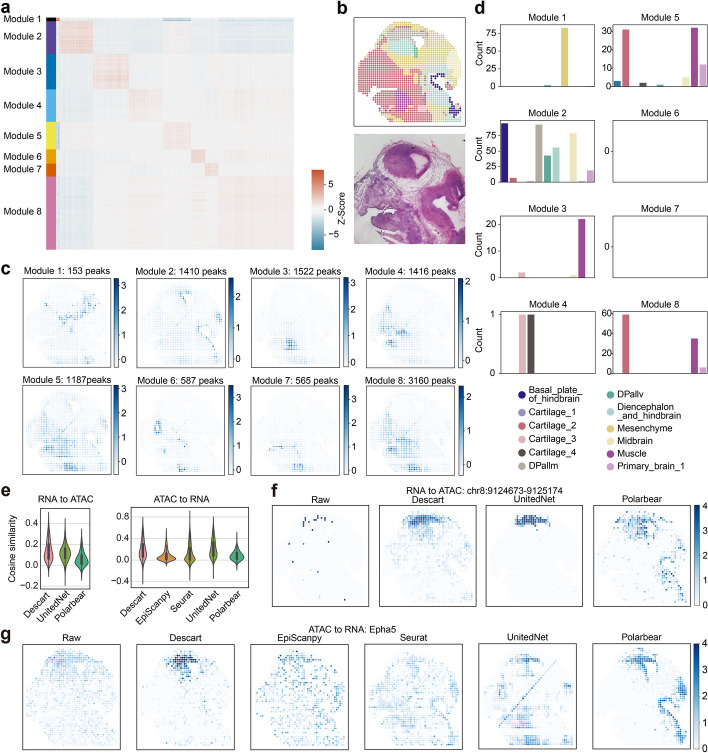
Spatial epigenomic technologies enable simultaneous capture of spatial location and chromatin accessibility of cells within tissue slices. Identifying peaks that display spatial variation and cellular heterogeneity is the key analytic task for characterizing the spatial chromatin accessibility landscape of complex tissues. Here, we propose an efficient and iterative model, Descart, for spatially variable peaks identification based on the graph of inter-cellular correlations. Through the comprehensive benchmarking, we demonstrate the superiority of Descart in revealing cellular heterogeneity and capturing tissue structure. Utilizing the graph of inter-cellular correlations, Descart shows its potential to denoise data, identify peak modules, and detect gene-peak interactions.
Keywords: Data imputation; Feature selection; Gene-peak interactions; Inter-cellular correlations; Peak module; Spatial ATAC-seq; Spatially variable peak.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that they have no competing interests.
Figures




References
-
- Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, Wardell E, Custodio J, Reimegard J, Salmen F, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(1647–1660): e1619. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
