

PharmRL: pharmacophore elucidation with deep geometric reinforcement learning
- PMID: 39736736
- PMCID: PMC11687028
- DOI: 10.1186/s12915-024-02096-5
PharmRL: pharmacophore elucidation with deep geometric reinforcement learning
Abstract
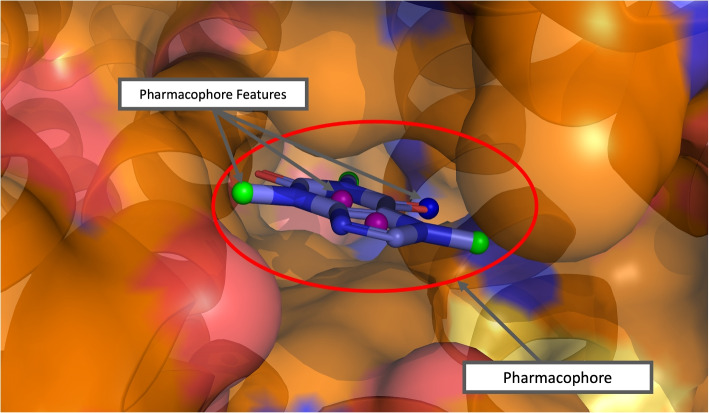
Background: Molecular interactions between proteins and their ligands are important for drug design. A pharmacophore consists of favorable molecular interactions in a protein binding site and can be utilized for virtual screening. Pharmacophores are easiest to identify from co-crystal structures of a bound protein-ligand complex. However, designing a pharmacophore in the absence of a ligand is a much harder task.
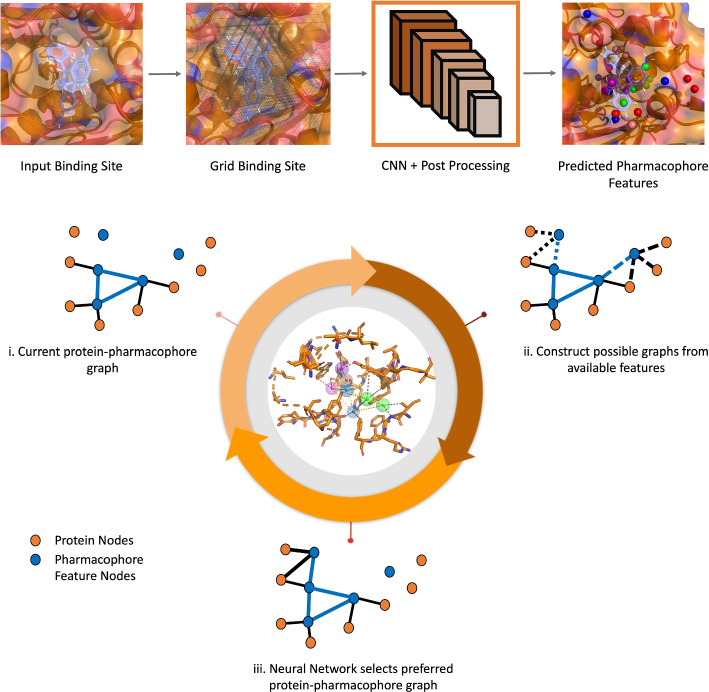
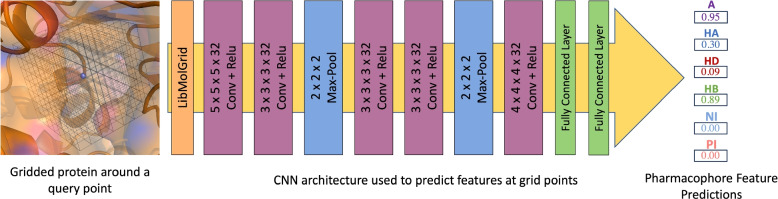
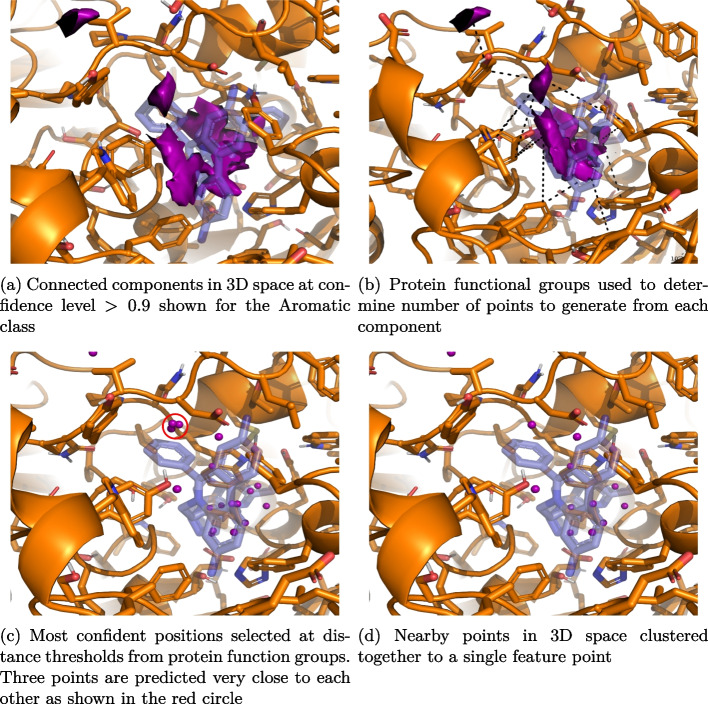
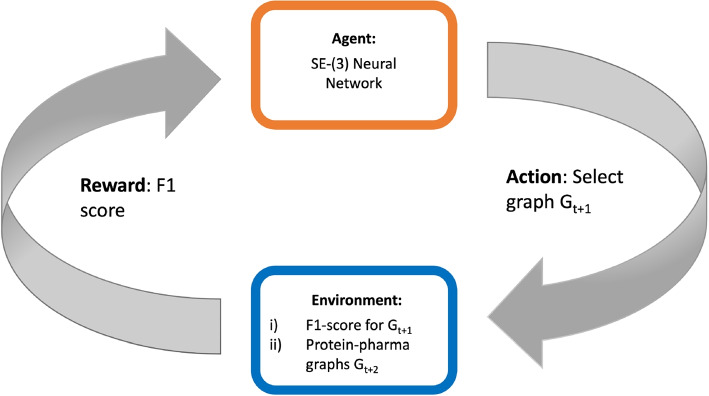
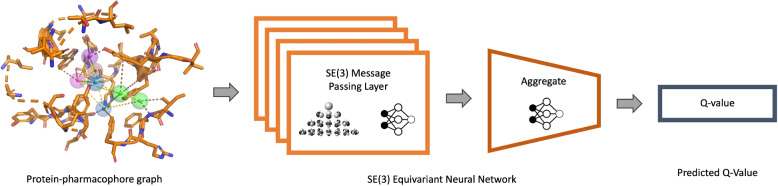
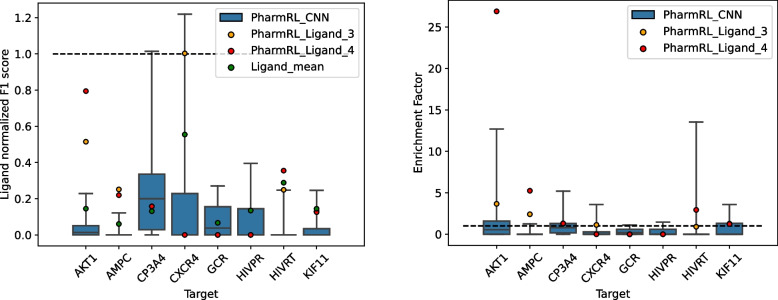
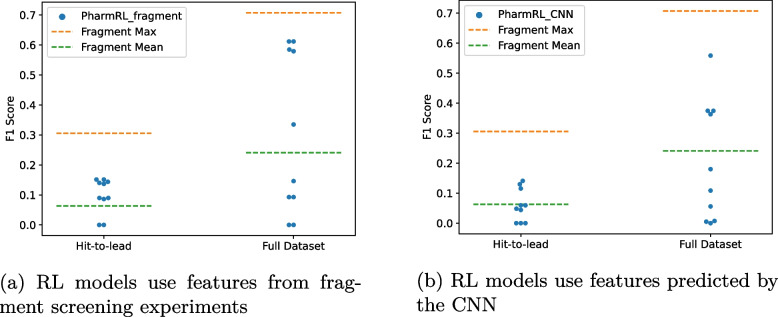
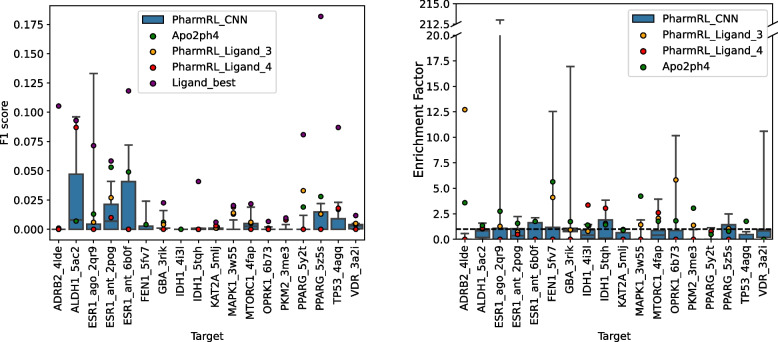
Results: In this work, we develop a deep learning method that can identify pharmacophores in the absence of a ligand. Specifically, we train a CNN model to identify potential favorable interactions in the binding site, and develop a deep geometric Q-learning algorithm that attempts to select an optimal subset of these interaction points to form a pharmacophore. With this algorithm, we show better prospective virtual screening performance, in terms of F1 scores, on the DUD-E dataset than random selection of ligand-identified features from co-crystal structures. We also conduct experiments on the LIT-PCBA dataset and show that it provides efficient solutions for identifying active molecules. Finally, we test our method by screening the COVID moonshot dataset and show that it would be effective in identifying prospective lead molecules even in the absence of fragment screening experiments.
Conclusions: PharmRL addresses the need for automated methods in pharmacophore design, particularly in cases where a cognate ligand is unavailable. Experimental results demonstrate that PharmRL generates functional pharmacophores. Additionally, we provide a Google Colab notebook to facilitate the use of this method.
Keywords: Machine learning; Pharmacophores; Protein-ligand interactions; Virtual screening.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: None to declare. Competing interests: The authors declare no competing interests.
Figures

Update of
-
PharmRL: Pharmacophore elucidation with Deep Geometric Reinforcement Learning.Res Sq [Preprint]. 2024 Sep 24:rs.3.rs-5033986. doi: 10.21203/rs.3.rs-5033986/v1. Res Sq. 2024. Update in: BMC Biol. 2024 Dec 31;22(1):301. doi: 10.1186/s12915-024-02096-5. PMID: 39399689 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
