

Genetic Code Expansion: Recent Developments and Emerging Applications
- PMID: 39737807
- PMCID: PMC11758808
- DOI: 10.1021/acs.chemrev.4c00216
Genetic Code Expansion: Recent Developments and Emerging Applications
Abstract
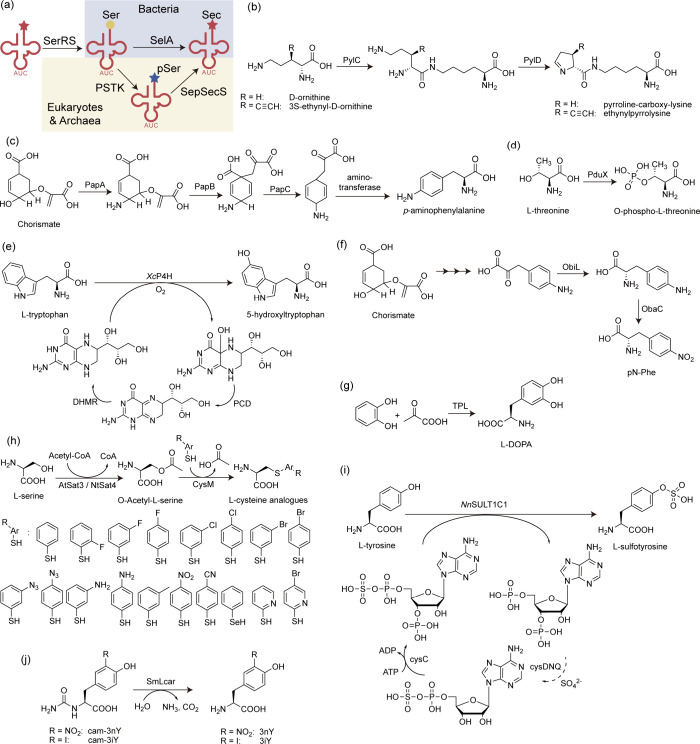
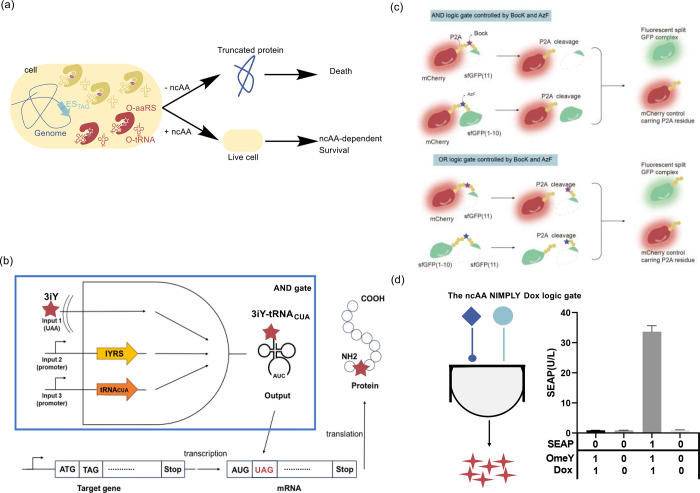
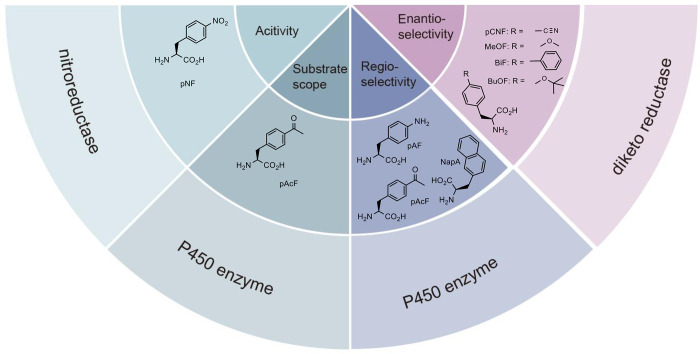
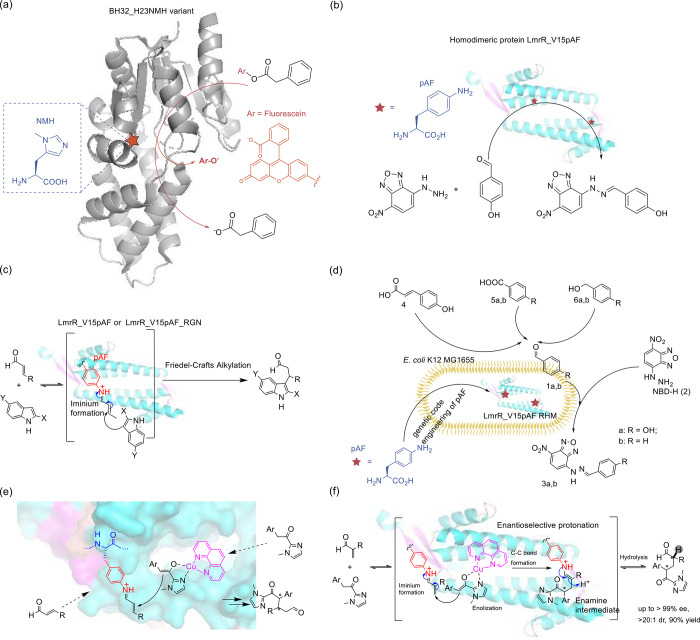
The concept of genetic code expansion (GCE) has revolutionized the field of chemical and synthetic biology, enabling the site-specific incorporation of noncanonical amino acids (ncAAs) into proteins, thus opening new avenues in research and applications across biology and medicine. In this review, we cover the principles of GCE, including the optimization of the aminoacyl-tRNA synthetase (aaRS)/tRNA system and the advancements in translation system engineering. Notable developments include the refinement of aaRS/tRNA pairs, enhancements in screening methods, and the biosynthesis of noncanonical amino acids. The applications of GCE technology span from synthetic biology, where it facilitates gene expression regulation and protein engineering, to medicine, with promising approaches in drug development, vaccine production, and gene editing. The review concludes with a perspective on the future of GCE, underscoring its potential to further expand the toolkit of biology and medicine. Through this comprehensive review, we aim to provide a detailed overview of the current state of GCE technology, its challenges, opportunities, and the frontier it represents in the expansion of the genetic code for novel biological research and therapeutic applications.
Conflict of interest statement
The authors declare the following competing financial interest(s): X.L. has financial interests in Demetrix and Synceres.
Figures
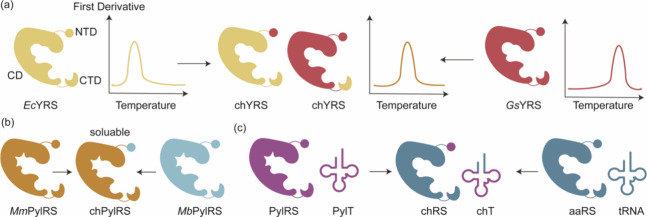
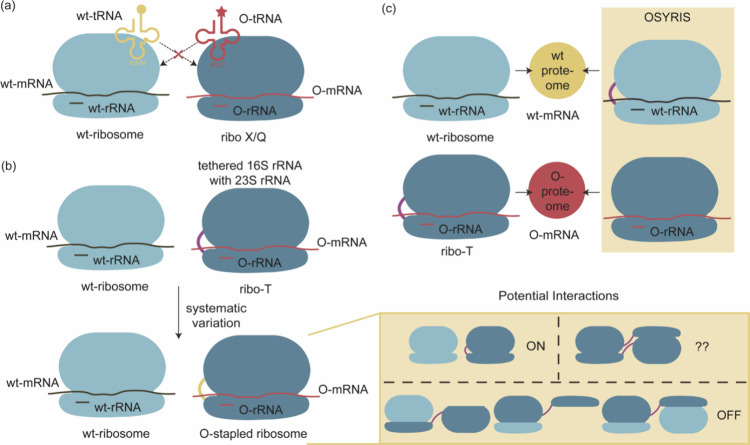
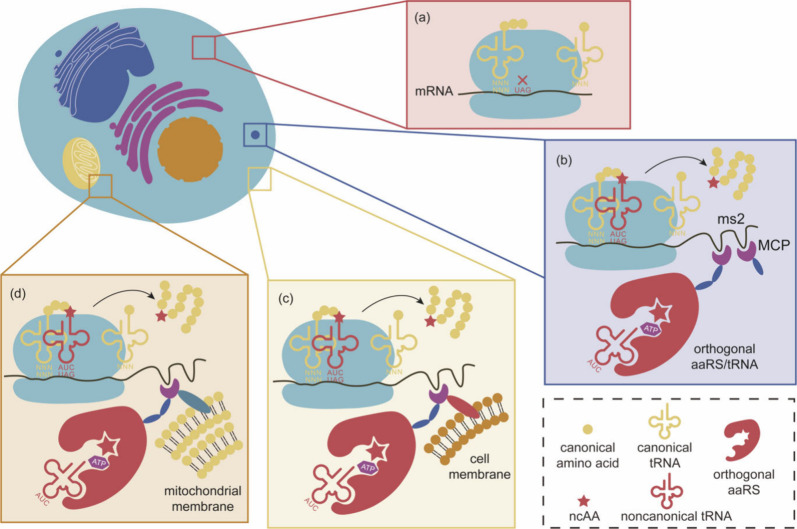
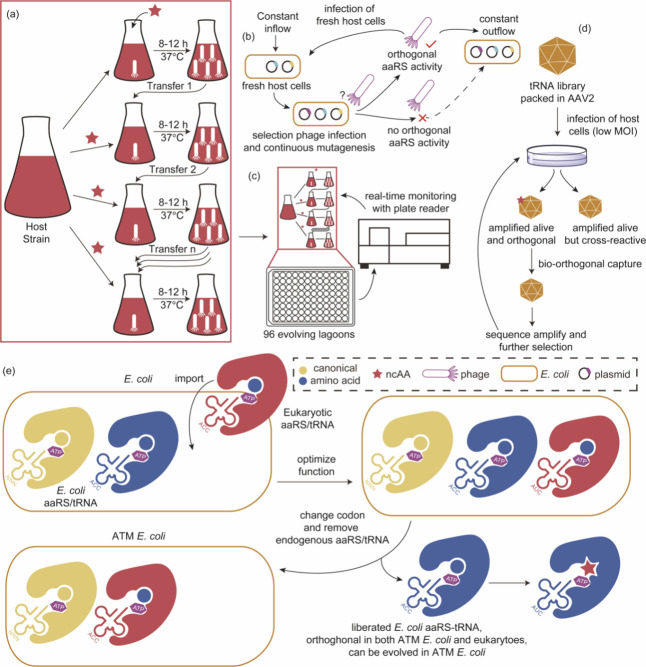
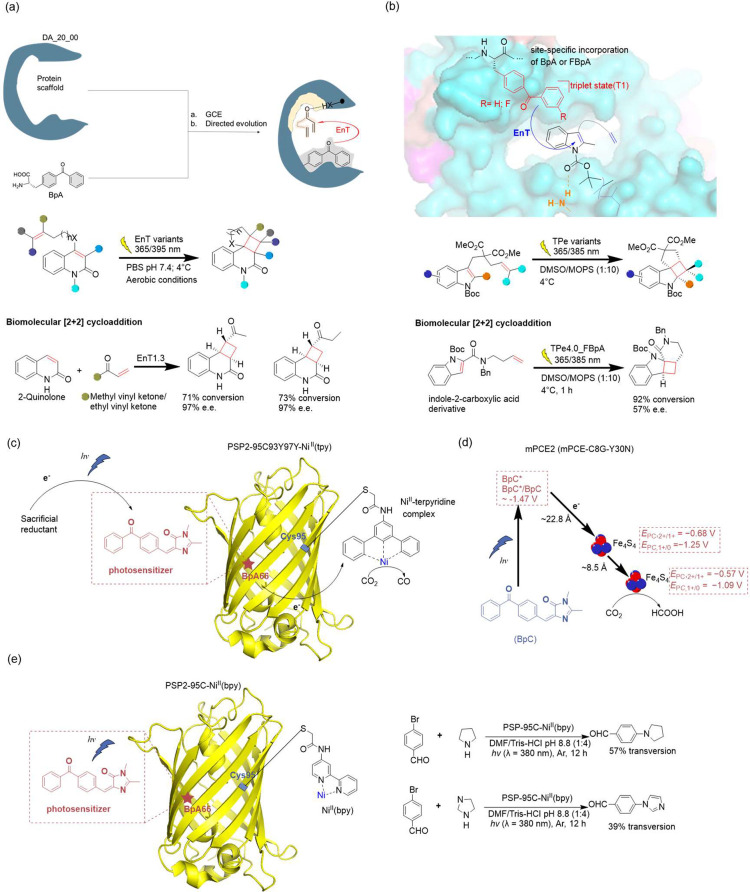
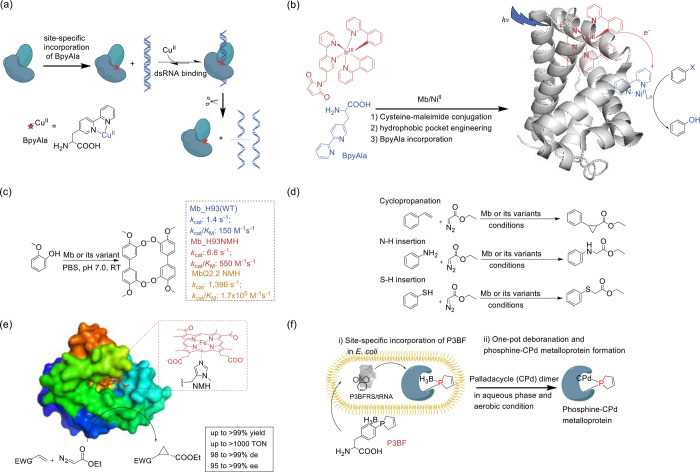
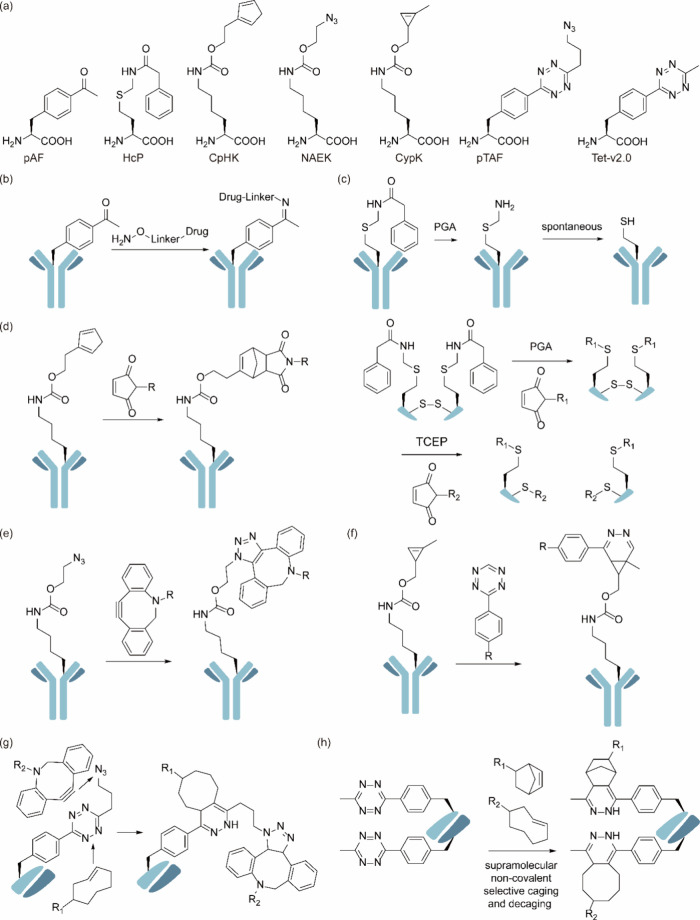
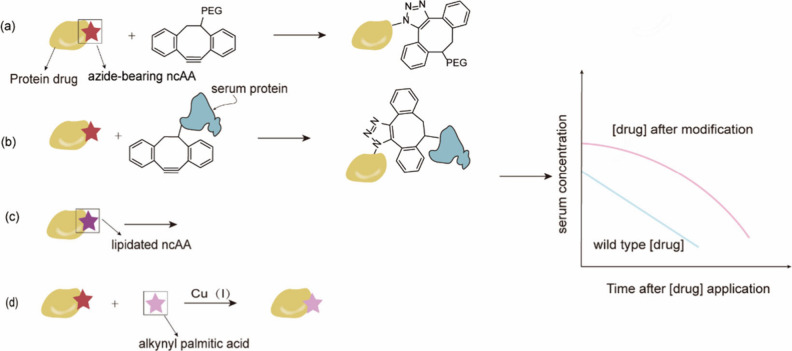
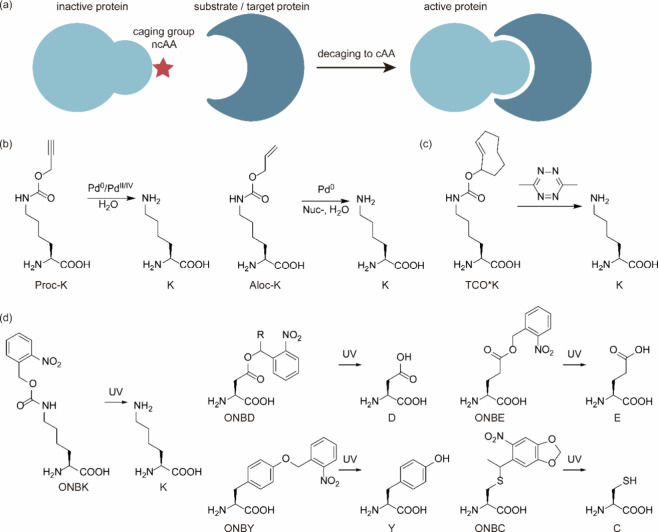
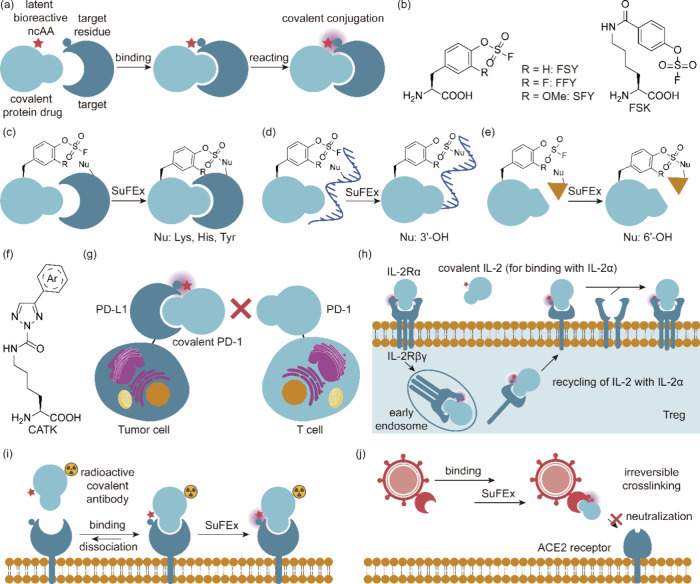
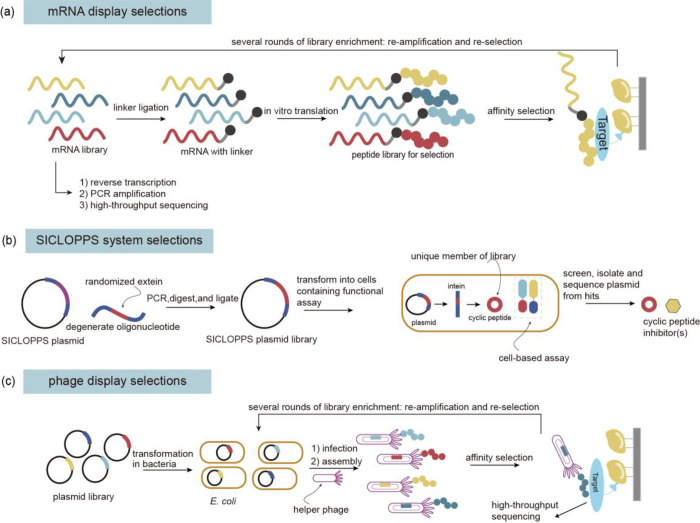
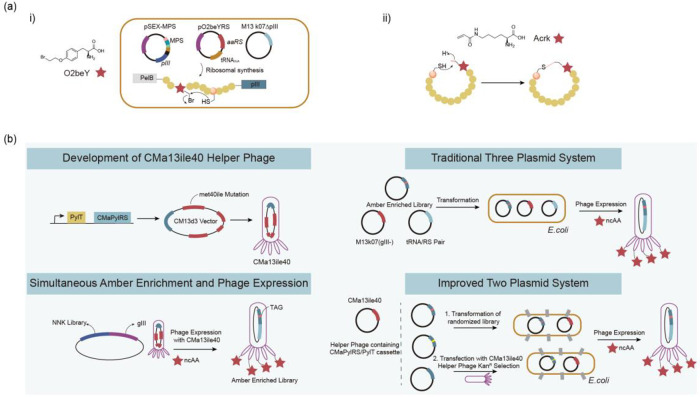
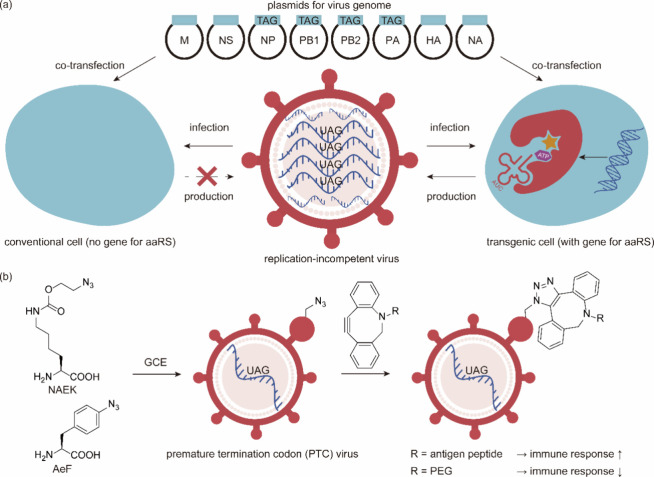
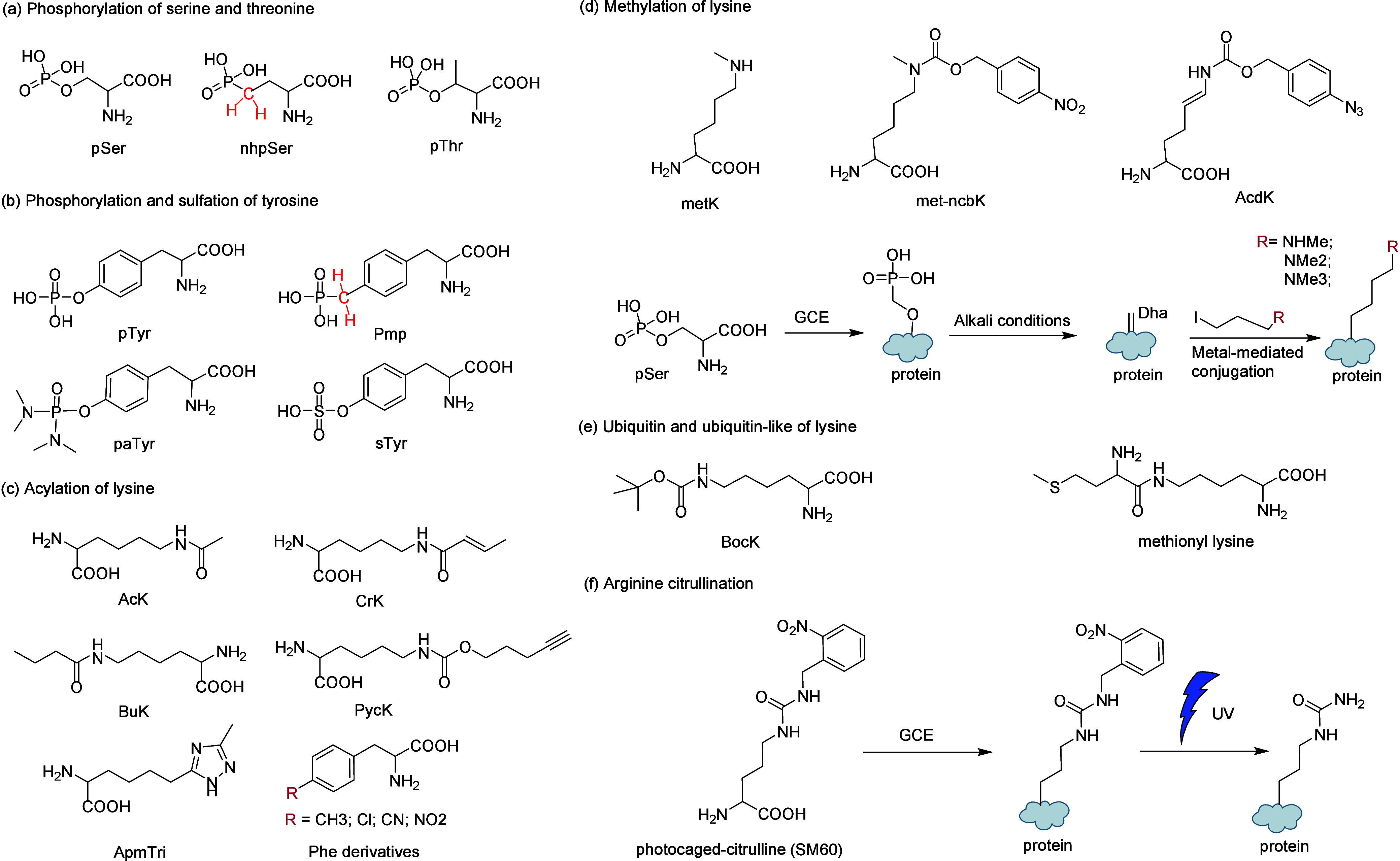
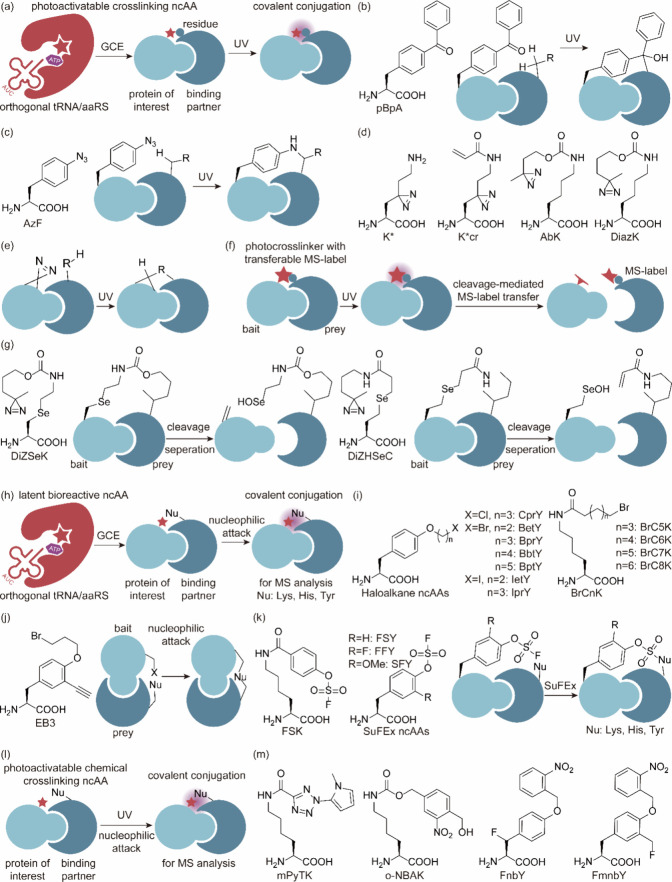
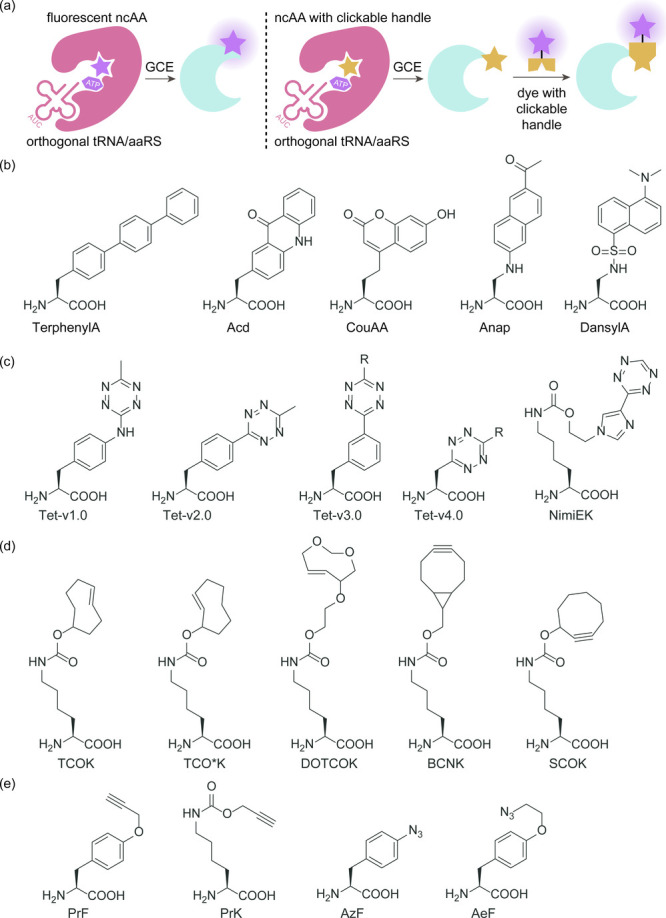
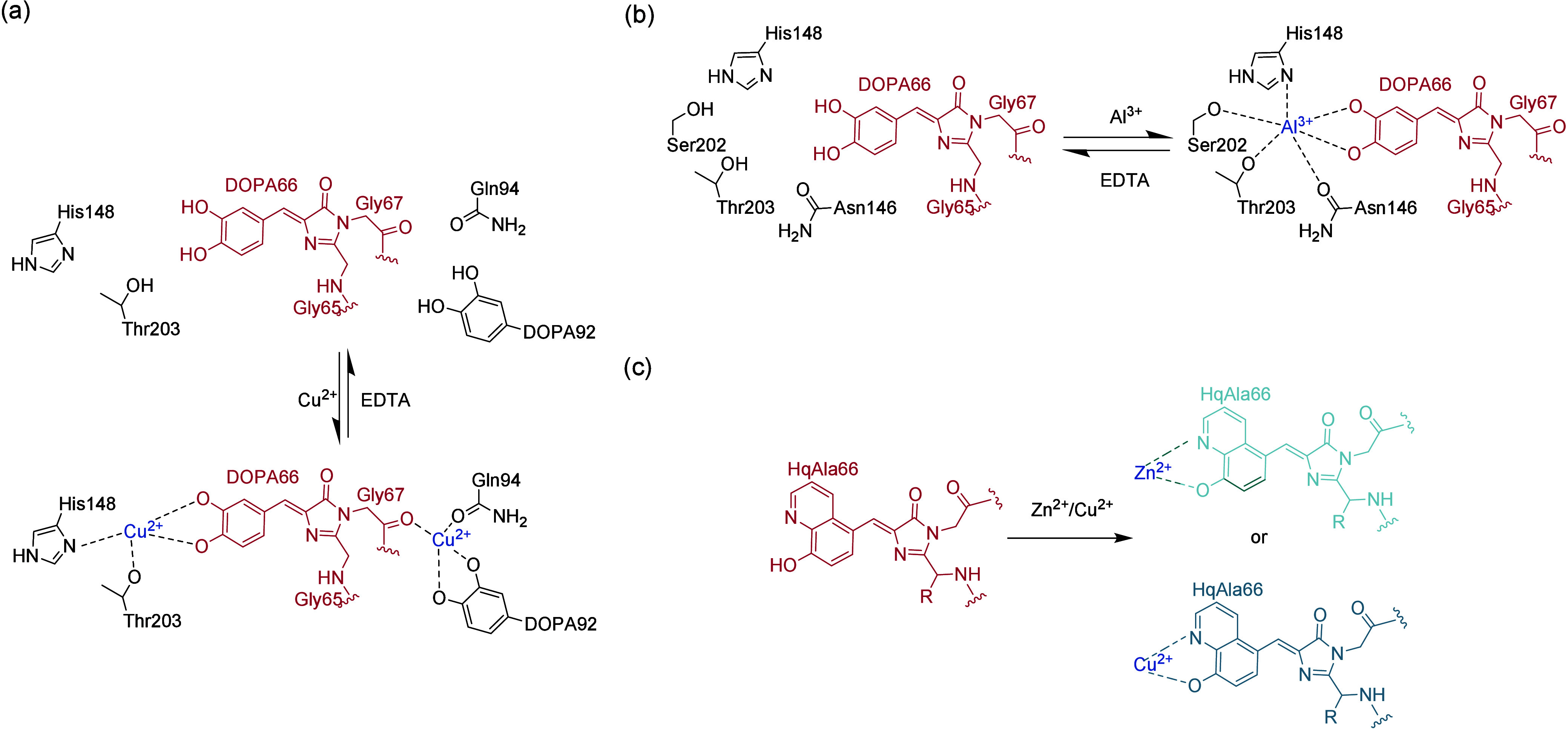
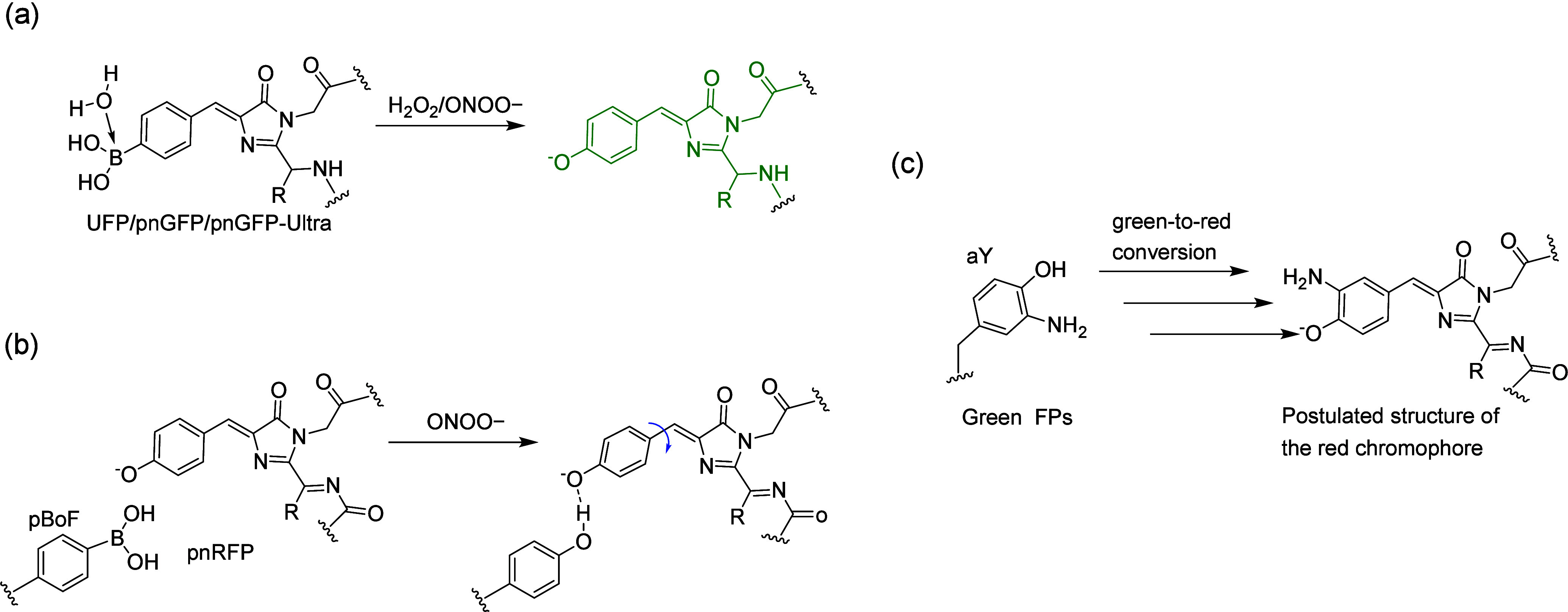
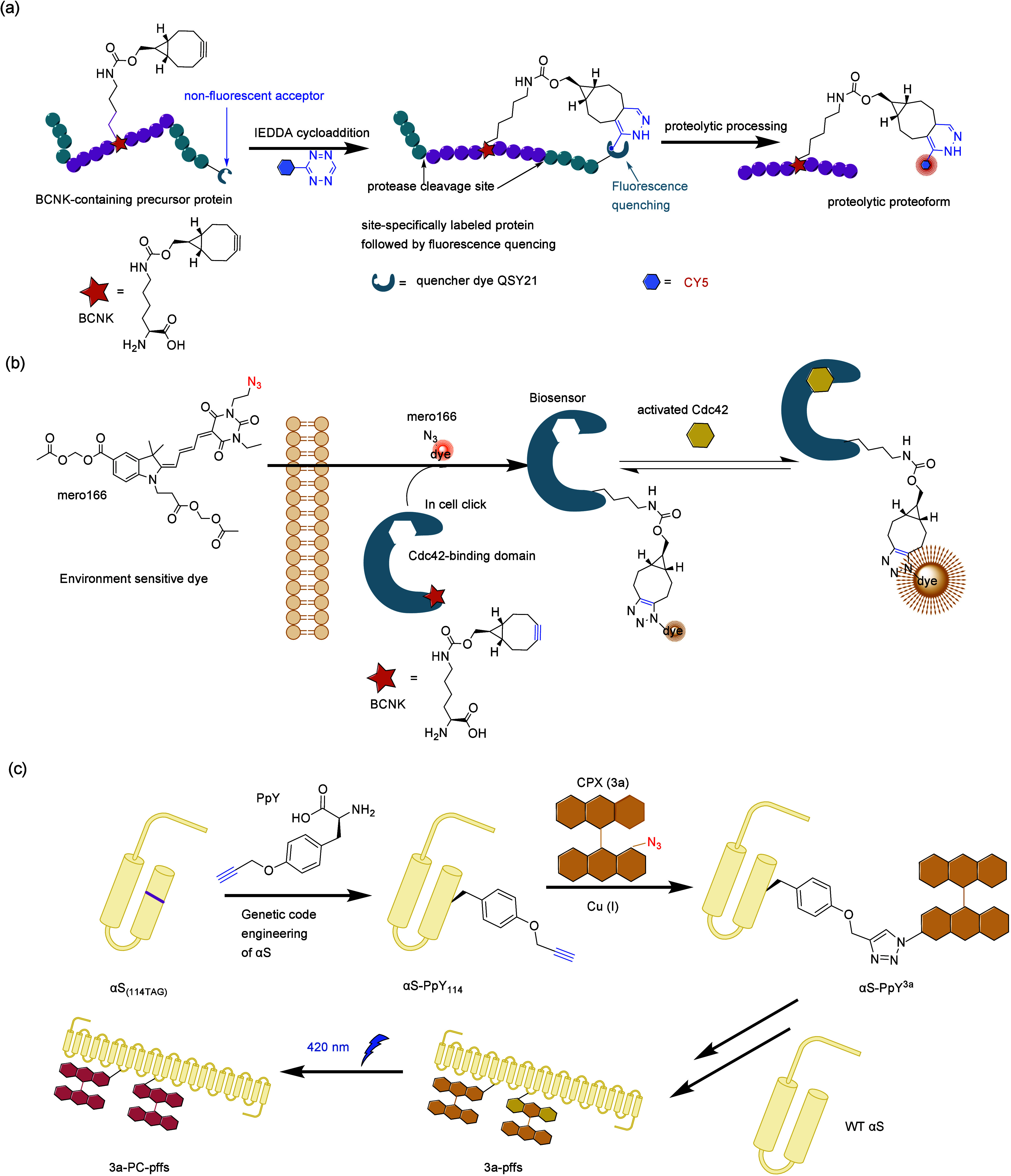
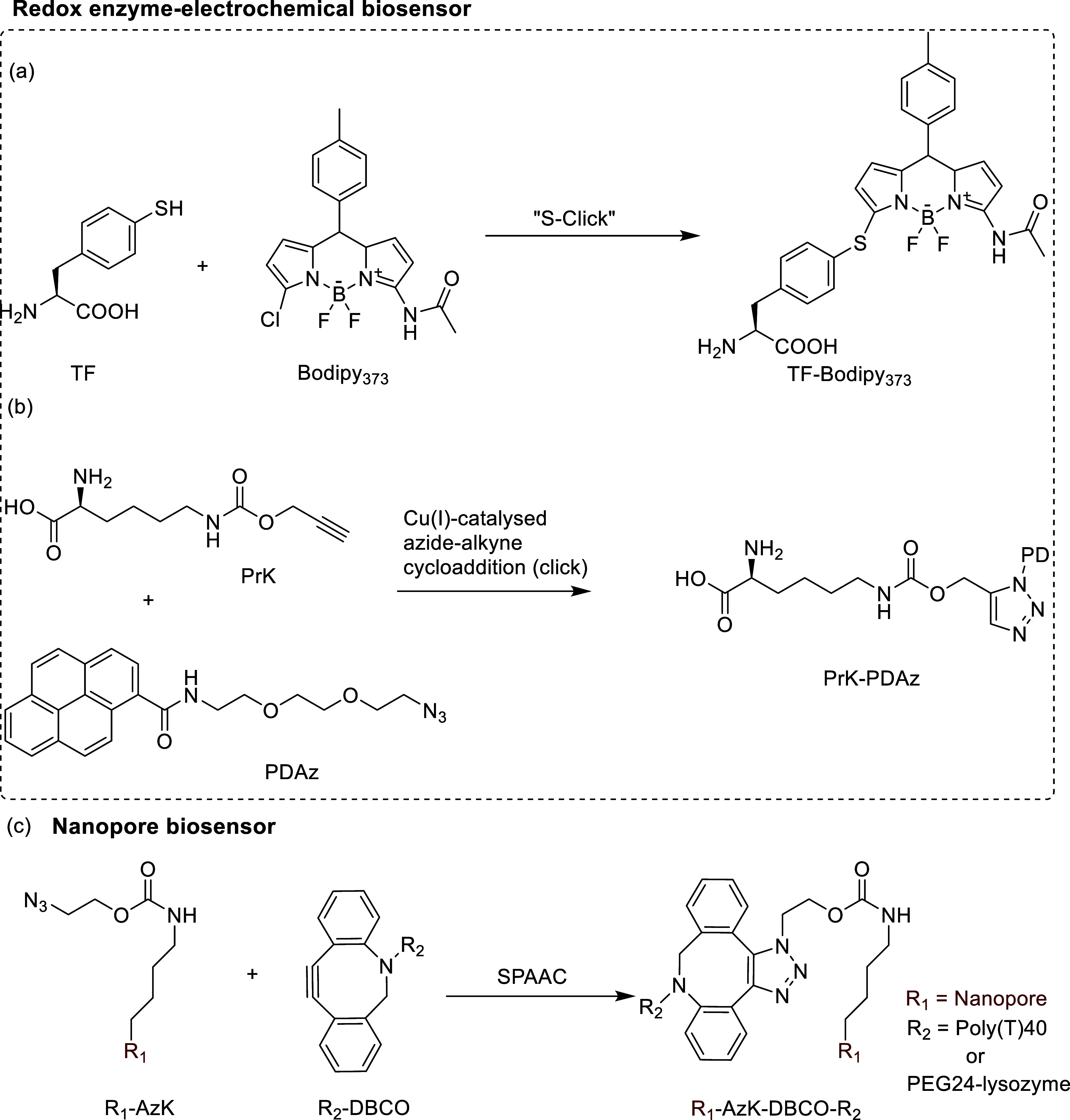
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
