

Haplotype-Resolved Genotyping and Association Analysis of 1,020 β-Thalassemia Patients by Targeted Long-Read Sequencing
- PMID: 39737841
- PMCID: PMC11884621
- DOI: 10.1002/advs.202410992
Haplotype-Resolved Genotyping and Association Analysis of 1,020 β-Thalassemia Patients by Targeted Long-Read Sequencing
Abstract
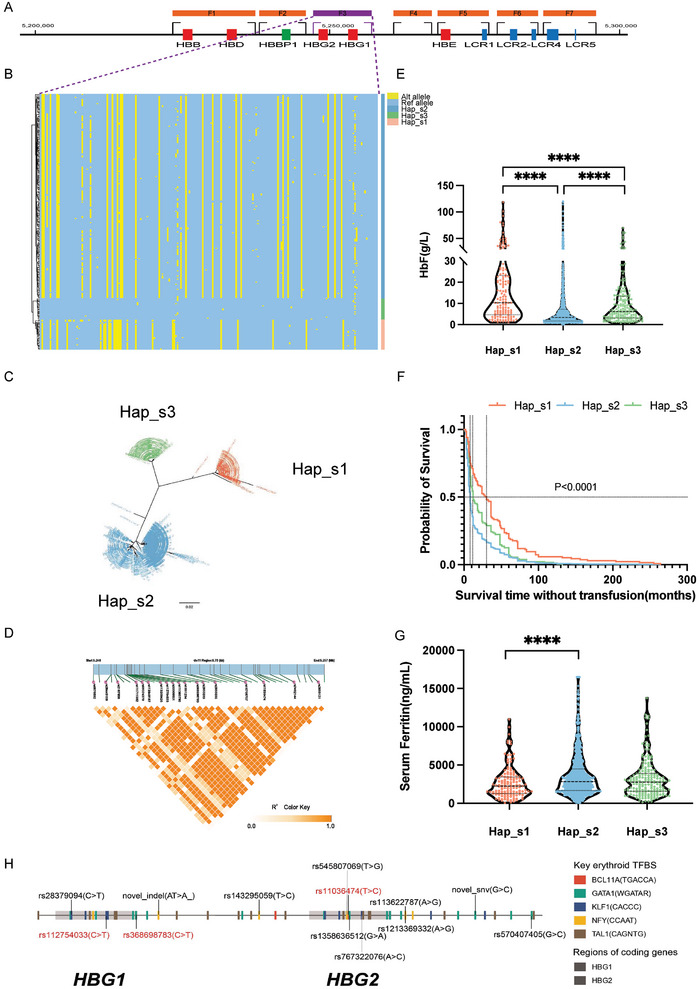
Despite the well-documented mutation spectra of β-thalassemia, the genetic variants and haplotypes of globin gene clusters modulating its clinical heterogeneity remain incompletely illustrated. Here, a targeted long-read sequencing (T-LRS) is demonstrated to capture 20 genes/loci in 1,020 β-thalassemia patients. This panel permits not only identification of thalassemia mutations at 100% of sensitivity and specificity, but also detection of rare structural variants (SVs) and single nucleotide variants (SNVs) in modifier genes/loci. The highly homologous regions of α-/β-globin gene clusters are then phased and 3 novel haplotypes in HBG1/HBG2 region are reported in this population of β-thalassemia patients. Furthermore, one of the haplotypes is associated with ameliorated symptoms of β-thalassemia. Similarly, 5 major haplotypes are identified in HBA1/HBA2 homologous region while one of them is found highly linked with deletional α-thalassemia mutations. Finally, rare mutations in erythroid transcription factors in DNMT1 and KLF1 associated with increased expression of fetal hemoglobin and reduced transfusion dependencies are identified. This study presents the largest T-LRS study for β-thalassemia patients to date, facilitating precise clinical diagnosis and haplotype phasing of globin gene clusters.
Keywords: fetal hemoglobin; thalassemia; third‐generation sequencing.
© 2024 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous