

MaizeCODE reveals bi-directionally expressed enhancers that harbor molecular signatures of maize domestication
- PMID: 39738013
- PMCID: PMC11685423
- DOI: 10.1038/s41467-024-55195-w
MaizeCODE reveals bi-directionally expressed enhancers that harbor molecular signatures of maize domestication
Erratum in
-
Author Correction: MaizeCODE reveals bi-directionally expressed enhancers that harbor molecular signatures of maize domestication.Nat Commun. 2025 Jul 23;16(1):6790. doi: 10.1038/s41467-025-62179-x. Nat Commun. 2025. PMID: 40702017 Free PMC article. No abstract available.
Abstract
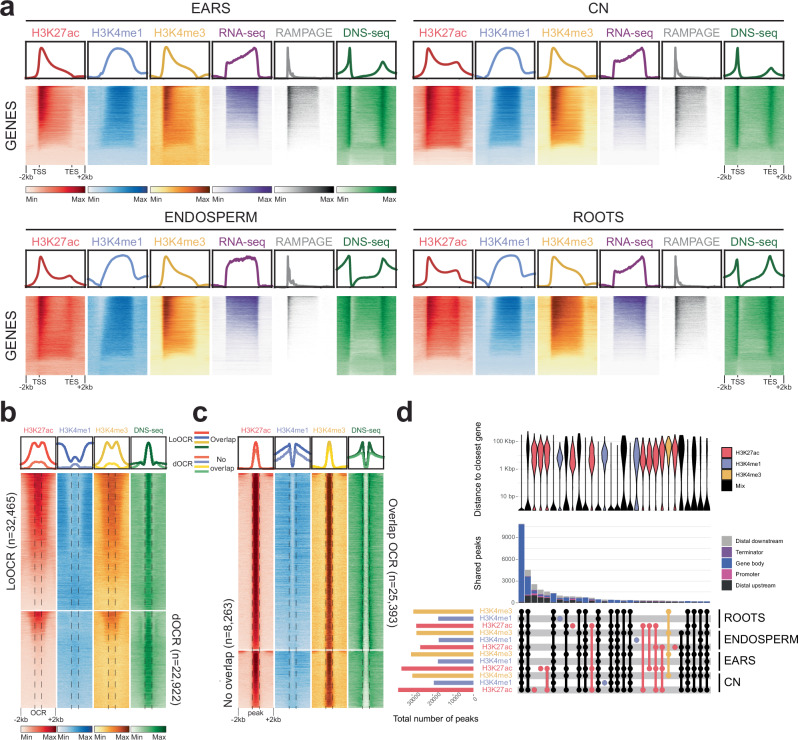
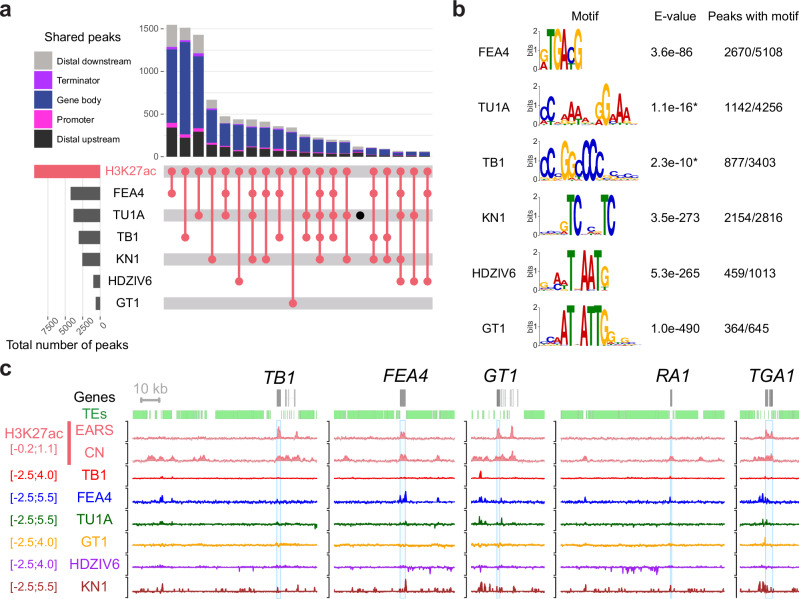
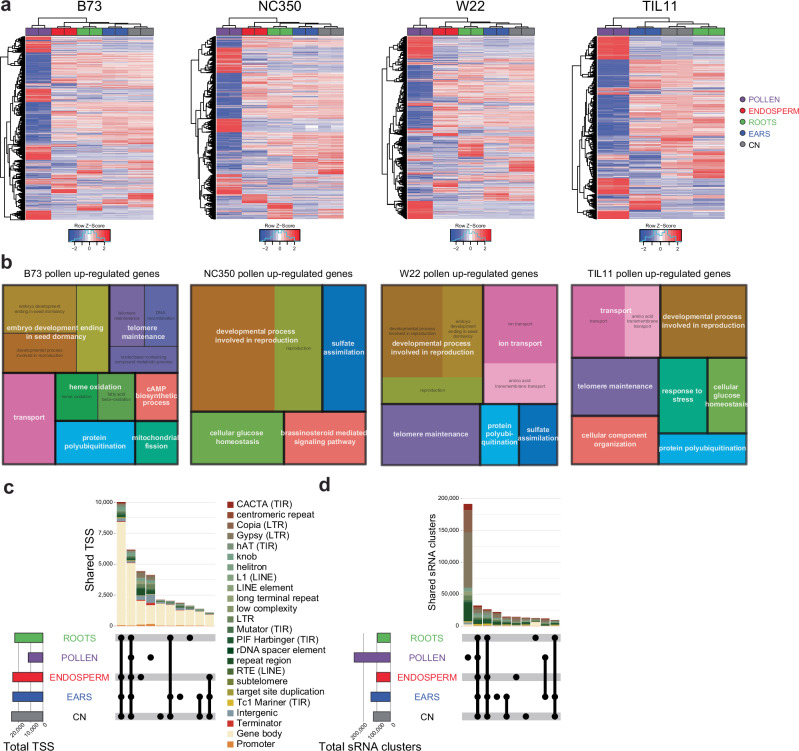
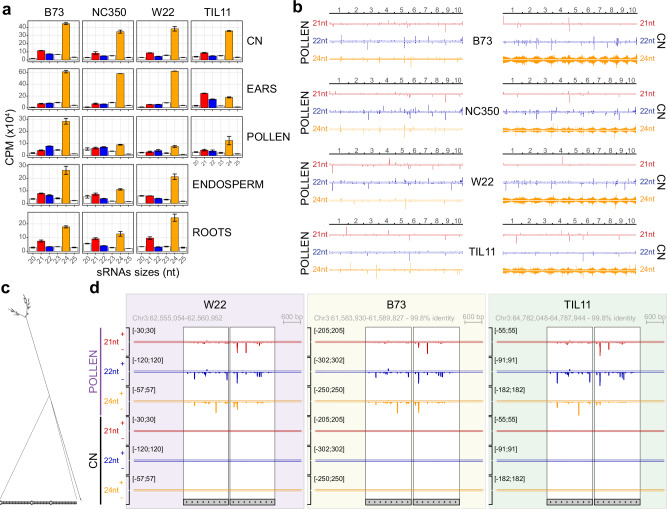
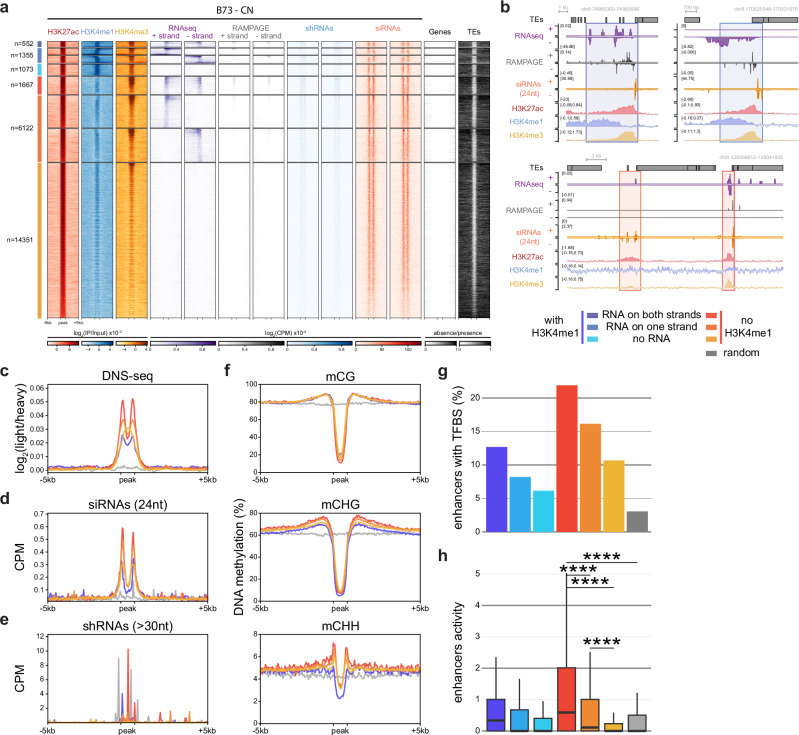
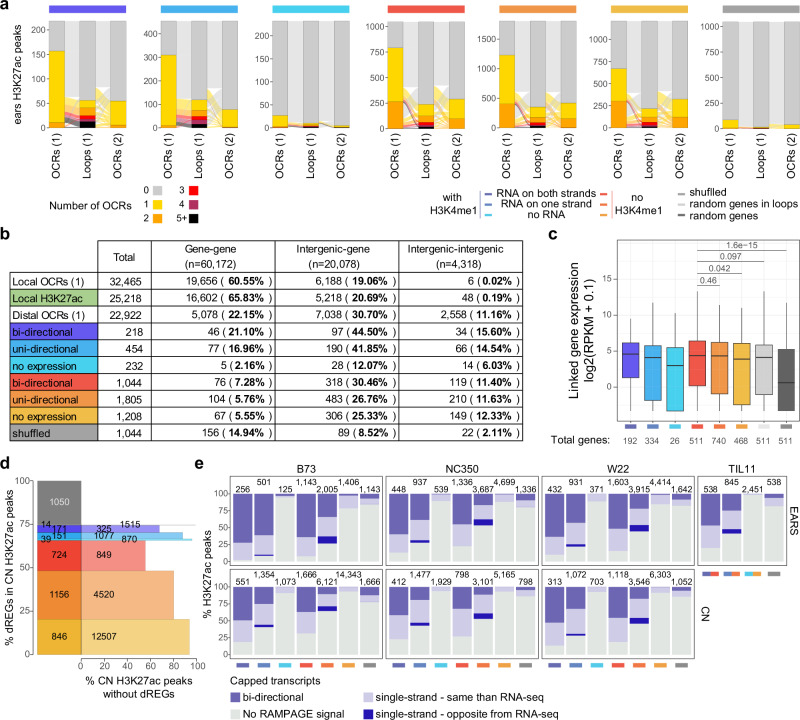
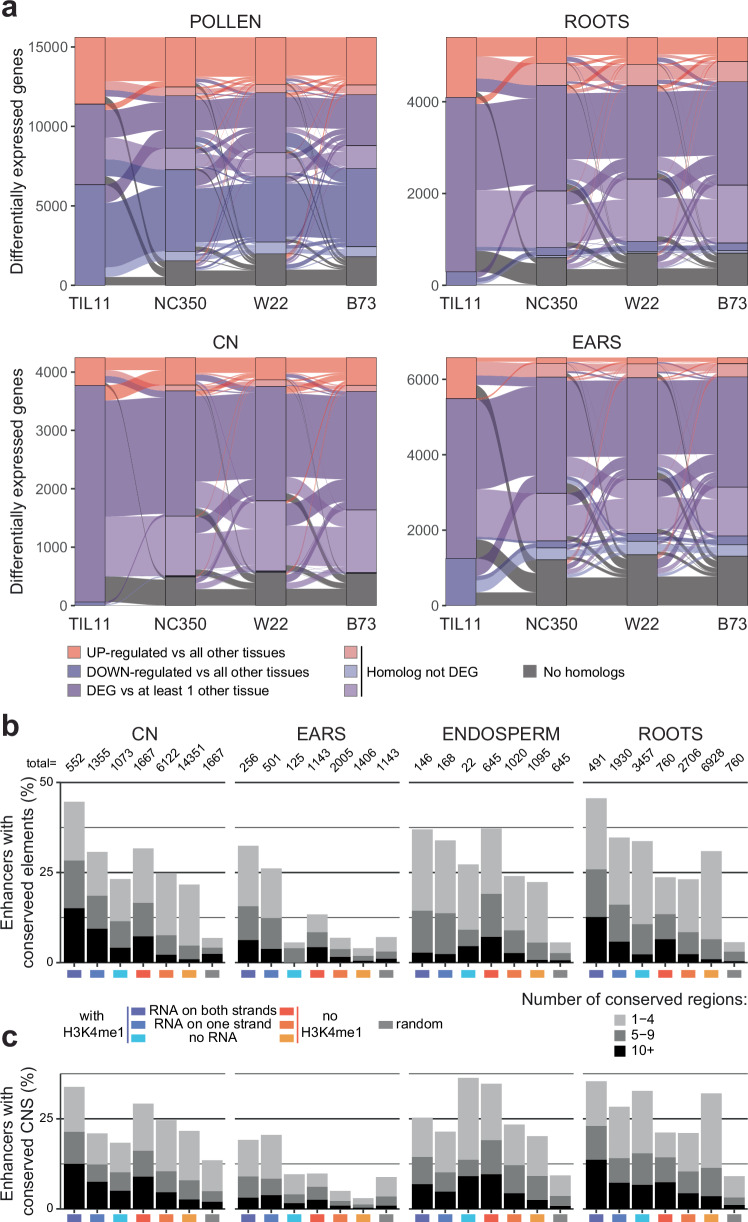
Modern maize (Zea mays ssp. mays) was domesticated from Teosinte parviglumis (Zea mays ssp. parviglumis), with subsequent introgressions from Teosinte mexicana (Zea mays ssp. mexicana), yielding increased kernel row number, loss of the hard fruit case and dissociation from the cob upon maturity, as well as fewer tillers. Molecular approaches have identified transcription factors controlling these traits, yet revealed that a complex regulatory network is at play. MaizeCODE deploys ENCODE strategies to catalog regulatory regions in the maize genome, generating histone modification and transcription factor ChIP-seq in parallel with transcriptomics datasets in 5 tissues of 3 inbred lines which span the phenotypic diversity of maize, as well as the teosinte inbred TIL11. Transcriptomic analysis reveals that pollen grains share features with endosperm, and express dozens of "proto-miRNAs" potential vestiges of gene drive and hybrid incompatibility. Integrated analysis with chromatin modifications results in the identification of a comprehensive set of regulatory regions in each tissue of each inbred, and notably of distal enhancers expressing non-coding enhancer RNAs bi-directionally, reminiscent of "super enhancers" in animal genomes. Furthermore, the morphological traits selected during domestication are recapitulated, both in gene expression and within regulatory regions containing enhancer RNAs, while highlighting the conflict between enhancer activity and silencing of the neighboring transposable elements.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
-
- Stitzer, M. C. & Ross-Ibarra, J. Maize domestication and gene interaction. N. Phytol.220, 395–408 (2018). - PubMed
-
- Wang, B. et al. Genome-wide selection and genetic improvement during modern maize breeding. Nat. Genet.52, 565–571 (2020). - PubMed
-
- Chen, L. et al. Genome sequencing reveals evidence of adaptive variation in the genus Zea. Nat. Genet.54, 1736–1745 (2022). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
