

Multivariate stochastic modeling for transcriptional dynamics with cell-specific latent time using SDEvelo
- PMID: 39738101
- PMCID: PMC11685993
- DOI: 10.1038/s41467-024-55146-5
Multivariate stochastic modeling for transcriptional dynamics with cell-specific latent time using SDEvelo
Abstract
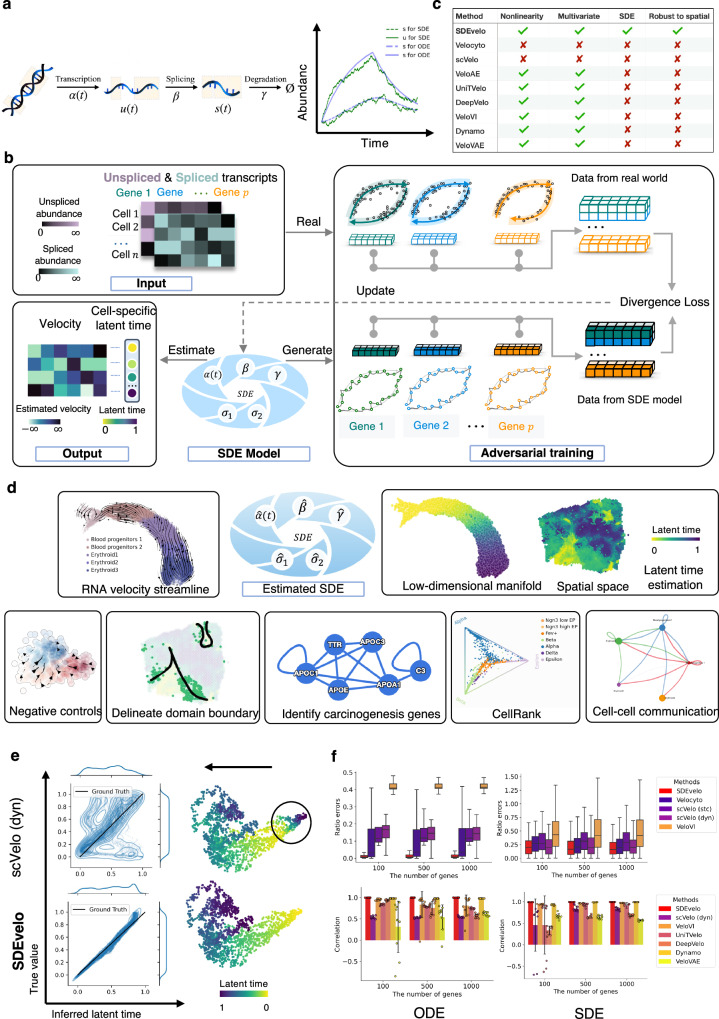
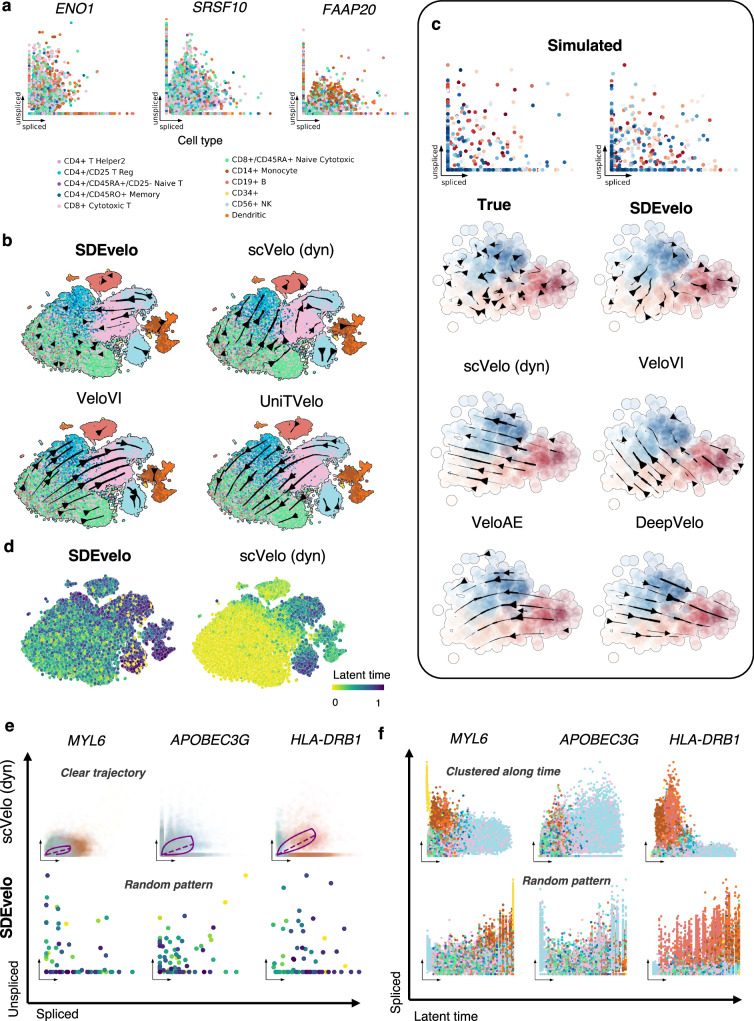
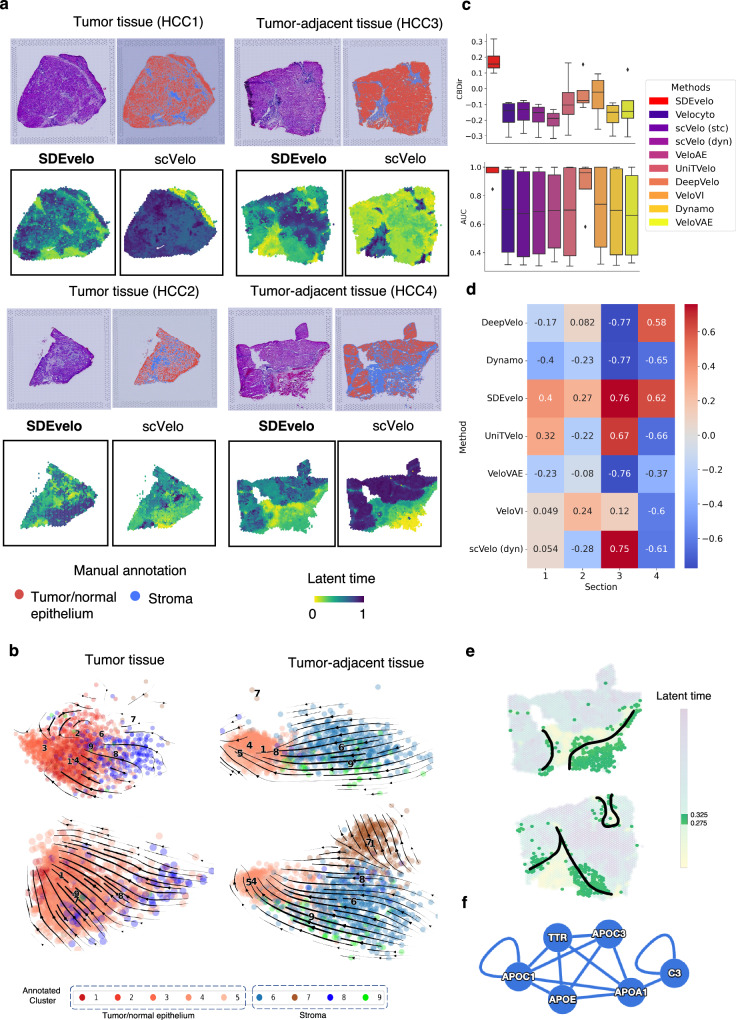
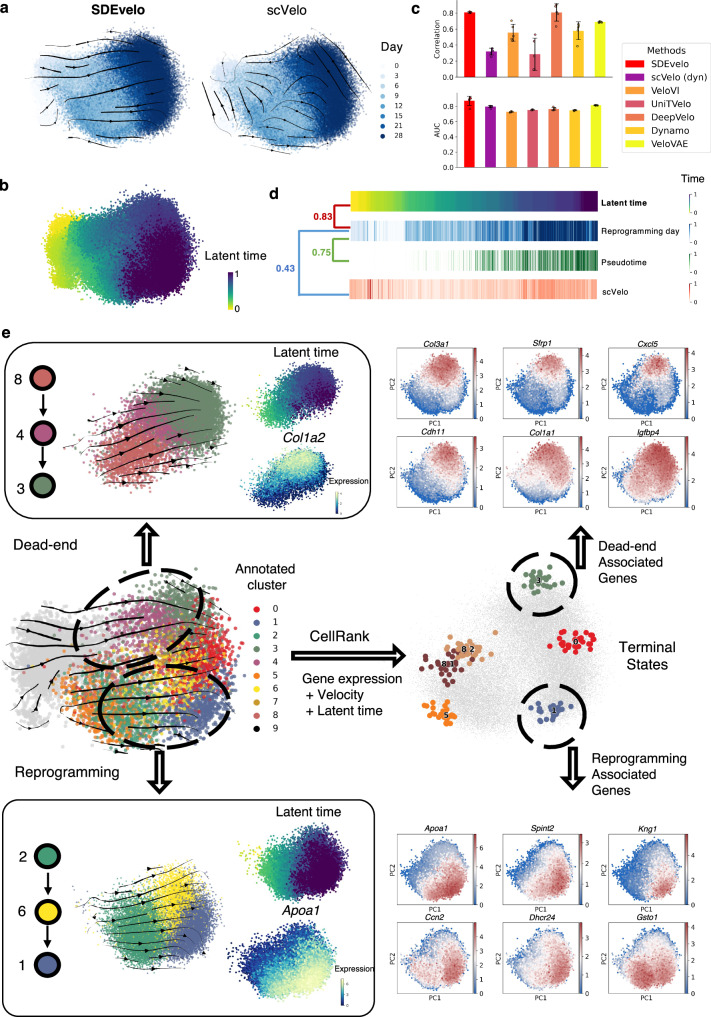
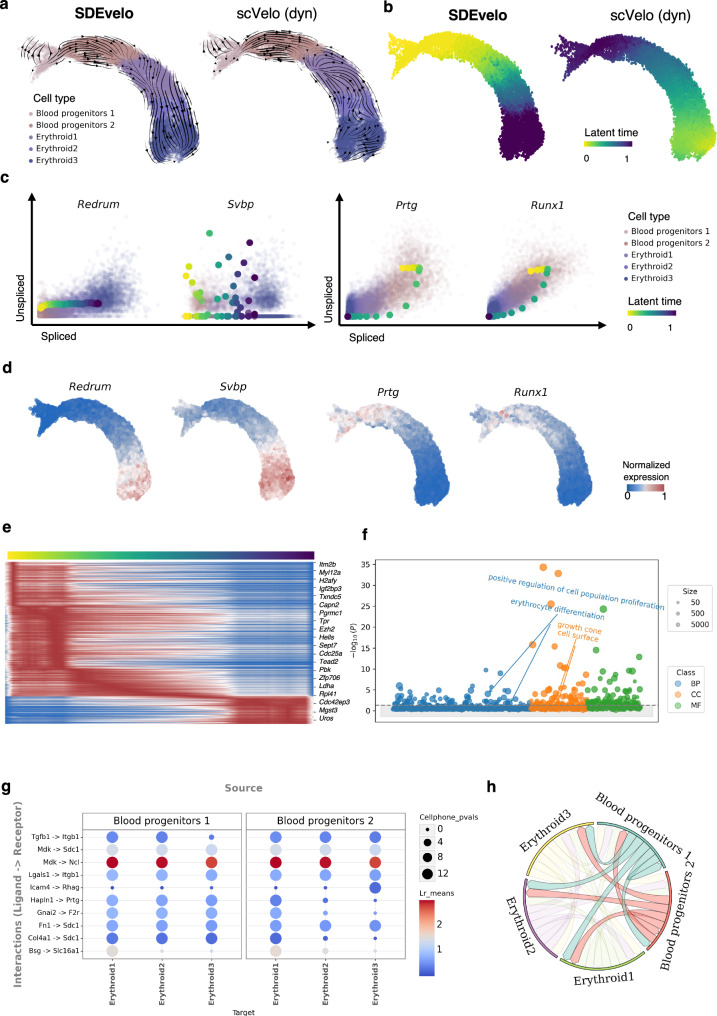
Recently, RNA velocity has driven a paradigmatic change in single-cell RNA sequencing (scRNA-seq) studies, allowing the reconstruction and prediction of directed trajectories in cell differentiation and state transitions. Most existing methods of dynamic modeling use ordinary differential equations (ODE) for individual genes without applying multivariate approaches. However, this modeling strategy inadequately captures the intrinsically stochastic nature of transcriptional dynamics governed by a cell-specific latent time across multiple genes, potentially leading to erroneous results. Here, we present SDEvelo, a generative approach to inferring RNA velocity by modeling the dynamics of unspliced and spliced RNAs via multivariate stochastic differential equations (SDE). Uniquely, SDEvelo explicitly models inherent uncertainty in transcriptional dynamics while estimating a cell-specific latent time across genes. Using both simulated and four scRNA-seq and spatial transcriptomics datasets, we show that SDEvelo can model the random dynamic patterns of mature-state cells while accurately detecting carcinogenesis. Additionally, the estimated gene-shared latent time can facilitate many downstream analyses for biological discovery. We demonstrate that SDEvelo is computationally scalable and applicable to both scRNA-seq and sequencing-based spatial transcriptomics data.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression.Nat Commun. 2015 Oct 22;6:8687. doi: 10.1038/ncomms9687. Nat Commun. 2015. PMID: 26489834 Free PMC article.
-
TSEE: an elastic embedding method to visualize the dynamic gene expression patterns of time series single-cell RNA sequencing data.BMC Genomics. 2019 Apr 4;20(Suppl 2):224. doi: 10.1186/s12864-019-5477-8. BMC Genomics. 2019. PMID: 30967106 Free PMC article.
-
A Guide to Trajectory Inference and RNA Velocity.Methods Mol Biol. 2023;2584:269-292. doi: 10.1007/978-1-0716-2756-3_14. Methods Mol Biol. 2023. PMID: 36495456
-
Temporal modelling using single-cell transcriptomics.Nat Rev Genet. 2022 Jun;23(6):355-368. doi: 10.1038/s41576-021-00444-7. Epub 2022 Jan 31. Nat Rev Genet. 2022. PMID: 35102309 Free PMC article. Review.
-
Integrating Dynamical Systems Modeling with Spatiotemporal scRNA-Seq Data Analysis.Entropy (Basel). 2025 Apr 22;27(5):453. doi: 10.3390/e27050453. Entropy (Basel). 2025. PMID: 40422408 Free PMC article. Review.
Cited by
-
Tet1 safeguards lineage allocation in intestinal stem cells.bioRxiv [Preprint]. 2025 May 13:2025.05.08.652522. doi: 10.1101/2025.05.08.652522. bioRxiv. 2025. PMID: 40463069 Free PMC article. Preprint.
References
-
- Mereu, E. et al. Benchmarking single-cell rna-sequencing protocols for cell atlas projects. Nat. Biotechnol.38, 747–755 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
