

Liver cancer multiomics reveals diverse protein kinase A disruptions convergently produce fibrolamellar hepatocellular carcinoma
- PMID: 39738196
- PMCID: PMC11685927
- DOI: 10.1038/s41467-024-55238-2
Liver cancer multiomics reveals diverse protein kinase A disruptions convergently produce fibrolamellar hepatocellular carcinoma
Abstract
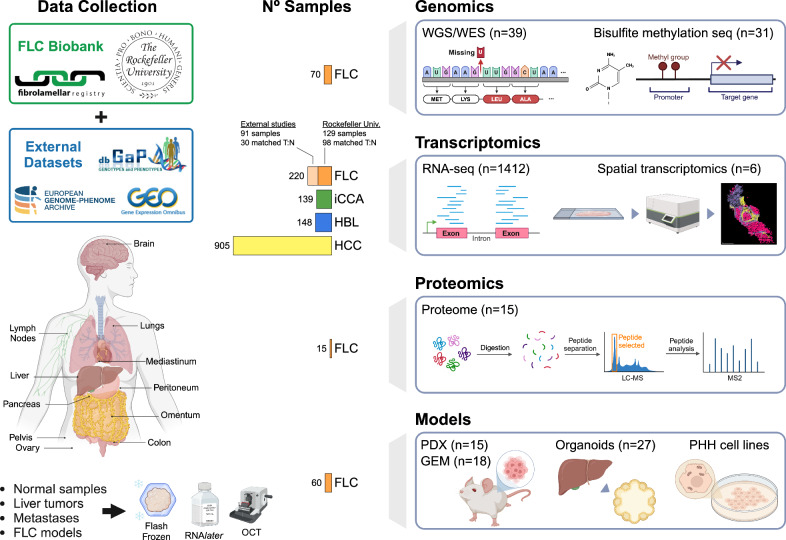
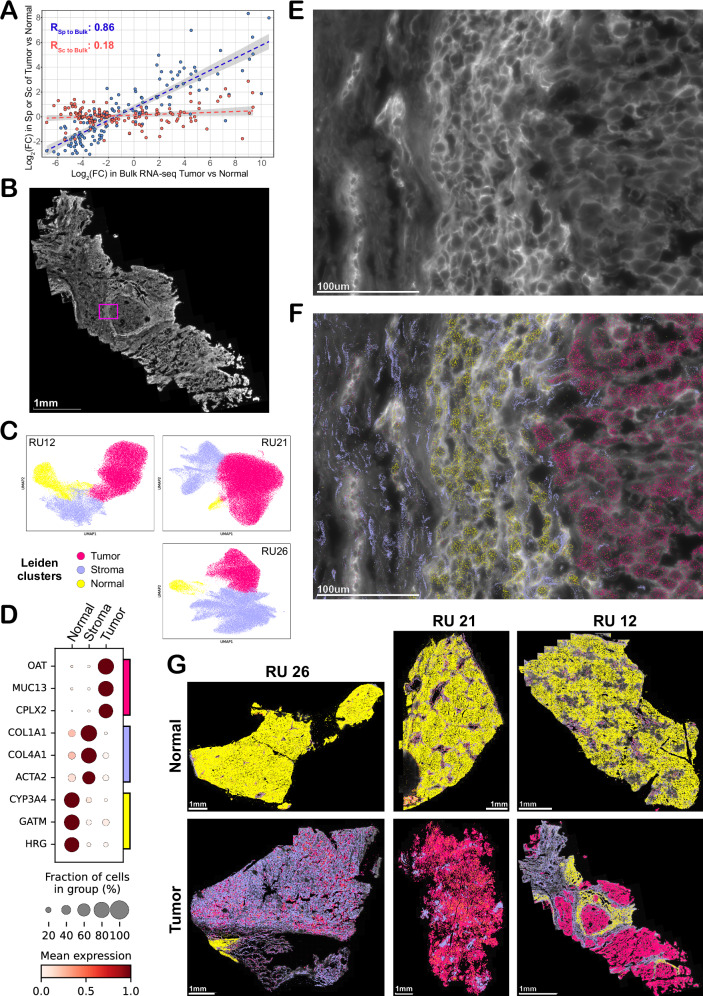
Fibrolamellar Hepatocellular Carcinoma (FLC) is a rare liver cancer characterized by a fusion oncokinase of the genes DNAJB1 and PRKACA, the catalytic subunit of protein kinase A (PKA). A few FLC-like tumors have been reported showing other alterations involving PKA. To better understand FLC pathogenesis and the relationships among FLC, FLC-like, and other liver tumors, we performed a massive multi-omics analysis. RNA-seq data of 1412 liver tumors from FLC, hepatocellular carcinoma, hepatoblastoma and intrahepatic cholangiocarcinoma are analyzed, obtaining transcriptomic signatures unrestricted by experimental processing methods. These signatures reveal which dysregulations are unique to specific tumors and which are common to all liver cancers. Moreover, the transcriptomic FLC signature identifies a unifying phenotype for all FLC tumors regardless of how PKA was activated. We study this signature at multi-omics and single-cell levels in the first spatial transcriptomic characterization of FLC, identifying the contribution of tumor, normal, stromal, and infiltrating immune cells. Additionally, we study FLC metastases, finding small differences from the primary tumors.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures






References
-
- Edmondson, H. A. Differential diagnosis of tumors and tumor-like lesions of liver in infancy and childhood. AMA J. Dis. Child91, 168–186 (1956). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
- P30 CA08748/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U54CA243126/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- UL1 TR001866/TR/NCATS NIH HHS/United States
- P50CA210964/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- 23CN18/Rally Foundation (Rally Foundation Inc.)
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
