

TRACERx analysis identifies a role for FAT1 in regulating chromosomal instability and whole-genome doubling via Hippo signalling
- PMID: 39738653
- PMCID: PMC11735399
- DOI: 10.1038/s41556-024-01558-w
TRACERx analysis identifies a role for FAT1 in regulating chromosomal instability and whole-genome doubling via Hippo signalling
Abstract
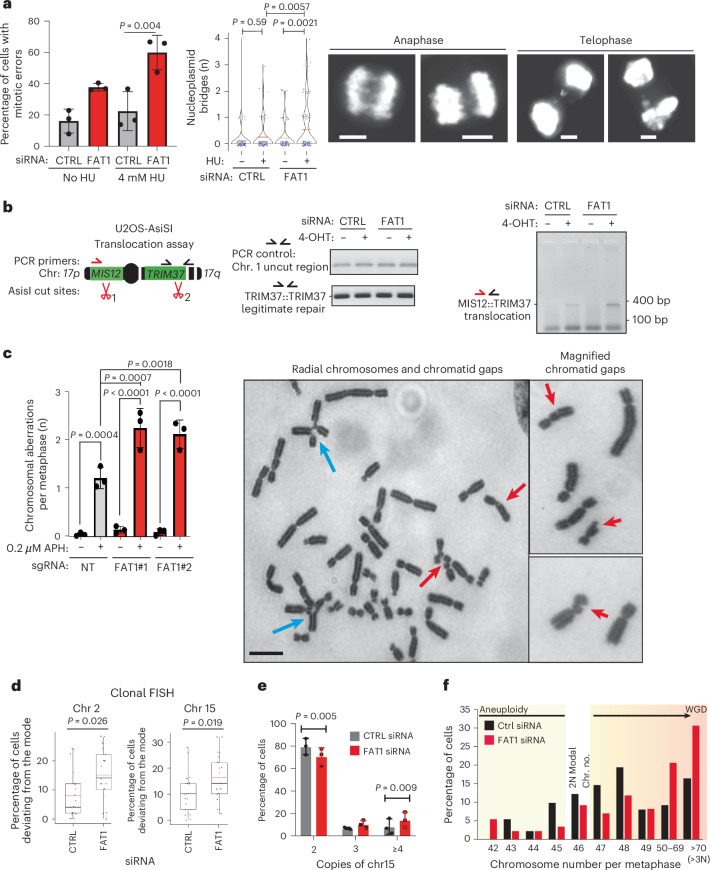
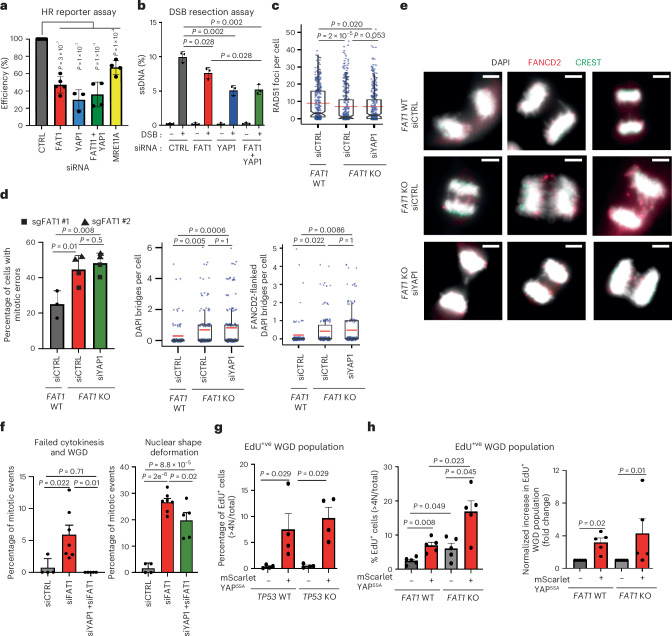
Chromosomal instability (CIN) is common in solid tumours and fuels evolutionary adaptation and poor prognosis by increasing intratumour heterogeneity. Systematic characterization of driver events in the TRACERx non-small-cell lung cancer (NSCLC) cohort identified that genetic alterations in six genes, including FAT1, result in homologous recombination (HR) repair deficiencies and CIN. Using orthogonal genetic and experimental approaches, we demonstrate that FAT1 alterations are positively selected before genome doubling and associated with HR deficiency. FAT1 ablation causes persistent replication stress, an elevated mitotic failure rate, nuclear deformation and elevated structural CIN, including chromosome translocations and radial chromosomes. FAT1 loss contributes to whole-genome doubling (a form of numerical CIN) through the dysregulation of YAP1. Co-depletion of YAP1 partially rescues numerical CIN caused by FAT1 loss but does not relieve HR deficiencies, nor structural CIN. Importantly, overexpression of constitutively active YAP15SA is sufficient to induce numerical CIN. Taken together, we show that FAT1 loss in NSCLC attenuates HR and exacerbates CIN through two distinct downstream mechanisms, leading to increased tumour heterogeneity.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: D.B. reports personal fees from NanoString and AstraZeneca and has a patent (PCT/GB2020/050221) issued on methods for cancer prognostication. M.A.B. has consulted for Achilles Therapeutics. M.J.-H. has received funding from CRUK, the National Institutes of Health National Cancer Institute, the International Association for the Study of Lung Cancer International Lung Cancer Foundation, the Lung Cancer Research Foundation, the Rosetrees Trust, the UK and Ireland Neuroendocrine Tumour Society and the NIHR. M.J.-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board (SAB) and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, Bristol Myers Squibb and Genentech. M.J.-H. is listed as a co-inventor on a European patent application relating to methods for the detection of lung cancer (PCT/US2017/028013). This patent has been licensed to commercial entities and, under the terms of their employment, M.J.-H. is due a share of any revenue generated from such license(s). M.J.-H. is also listed as a co-inventor on a GB priority patent application (GB2400424.4) with title ‘Treatment and prevention of lung cancer’. K.L. has a patent pending on indel burden and checkpoint inhibitor response, has received speaker fees from Roche Tissue Diagnostics and research funding from the CRUK Therapeutic Discovery Laboratories–Ono Pharmaceutical–LifeArc alliance and Genesis Therapeutics and has held consulting roles with Ellipses Pharma, Monopteros and Kynos Therapeutics. N.J.B. is listed as a co-inventor on a patent related to the identification of responders to cancer treatment (PCT/GB2018/051912), has submitted a patent application (PCT/GB2020/050221) on methods for cancer prognostication and has a patent on methods for predicting anti-cancer response (US14/466,208). N.M. has stock options in, and has consulted for, Achilles Therapeutics and holds a European patent in determining human leukocyte antigen (HLA) LOH (PCT/GB2018/052004), has a patent pending on determining HLA disruption (PCT/EP2023/059039) and is a co-inventor on a patent on the identification of responders to cancer treatment (PCT/GB2018/051912). N.K. acknowledges grants from AstraZeneca. C.S. acknowledges grants from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Pfizer, Roche VENTANA, Invitae (previously ArcherDX; a collaboration on minimal residual disease sequencing technologies), Ono Pharmaceutical and Personalis. He is Chief Investigator for the AZ MeRmaiD-1 and -2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri Trial funded by GRAIL and a paid member of GRAIL’s SAB. He receives consultant fees from Achilles Therapeutics (and is also a SAB member), Bicycle Therapeutics (and is also a SAB member), Genentech, Medicxi, the China Innovation Centre of Roche (formerly the Roche Innovation Centre – Shanghai); Metabomed, until July 2022, Relay Therapeutics (and is also a SAB member), SAGA Diagnostics (and is also a SAB member) and the Sarah Cannon Research Institute. C.S. has received honoraria from Amgen, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer and Roche VENTANA. C.S. has previously held stock options in ApoGen Biotechnologies and GRAIL and currently has stock options in EPIC Bioscience, Bicycle Therapeutics, Relay Therapeutics and Achilles Therapeutics. C.S. is also a co-founder of Achilles Therapeutics. C.S. declares patents and patent applications relating to methods for the detection of lung cancer (PCT/US2017/028013), neoantigen targeting (PCT/EP2016/059401), the identification of patent response to immune checkpoint blockade (PCT/EP2016/071471), the identification of patients who respond to cancer treatment (PCT/GB2018/051912), the determination of HLA LOH (PCT/GB2018/052004), the prediction of survival rates of patients with cancer (PCT/GB2020/050221) and methods and systems for tumour monitoring (PCT/EP2022/077987). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumour recurrence. This patent has been licensed to a commercial entity and, under the terms of their employment, C.S. is due a share of any revenue generated from such license(s). The remaining authors declare no competing interests.
Figures
















References
-
- Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med.376, 2109–2121 (2017). - PubMed
MeSH terms
Substances
Grants and funding
- C416/A21999/Cancer Research UK (CRUK)
- 211179/Z/18/Z/Wellcome Trust (Wellcome)
- BCRF 23-157/Breast Cancer Research Foundation (BCRF)
- MR/V033077/1/RCUK | Medical Research Council (MRC)
- ID16584/Novo Nordisk Fonden (Novo Nordisk Foundation)
- AUFF-E-2018-7-14/Aarhus Universitets Forskningsfond (Aarhus University Research Foundation)
- EDDPMA-Nov21/100034/Cancer Research UK (CRUK)
- NNF21OC0071483/Novo Nordisk Fonden (Novo Nordisk Foundation)
- PROTEUS/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- C11496/A30025/Cancer Research UK (CRUK)
- RP/EA/180007 & RF\ERE\231118/Royal Society
- C11496/A17786/Cancer Research UK (CRUK)
- BCRF-22-157/Breast Cancer Research Foundation (BCRF)
- NNF23OC0085954/Novo Nordisk Fonden (Novo Nordisk Foundation)
- 835297/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- M630/Rosetrees Trust
- R322-A17482/Kr& #x00E6;ftens Bek& #x00E6;mpelse (Danish Cancer Society)
- RSRP\R\210001/Royal Society
- R272-2017-4040/Lundbeckfonden (Lundbeck Foundation)
- CC2041/WT_/Wellcome Trust/United Kingdom
- NNF20OC0060590/Novo Nordisk Fonden (Novo Nordisk Foundation)
- C69256/A30194/Cancer Research UK (CRUK)
LinkOut - more resources
Full Text Sources
