

Endothelial cell-selective adhesion molecule deficiency exhibits increased pulmonary vascular resistance due to impaired endothelial nitric oxide signaling
- PMID: 39740345
- PMCID: PMC12269549
- DOI: 10.1152/ajpheart.00593.2024
Endothelial cell-selective adhesion molecule deficiency exhibits increased pulmonary vascular resistance due to impaired endothelial nitric oxide signaling
Abstract
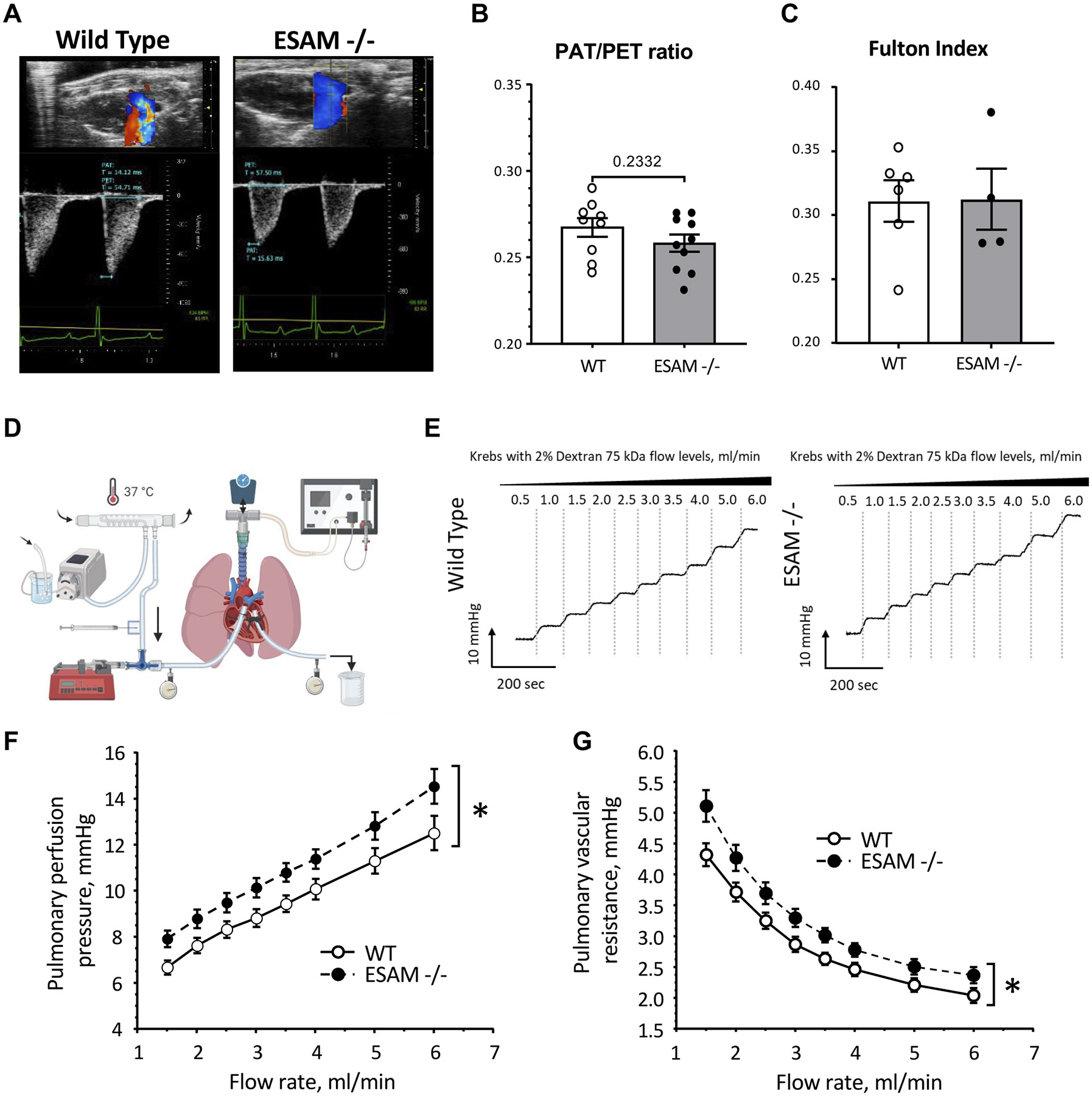
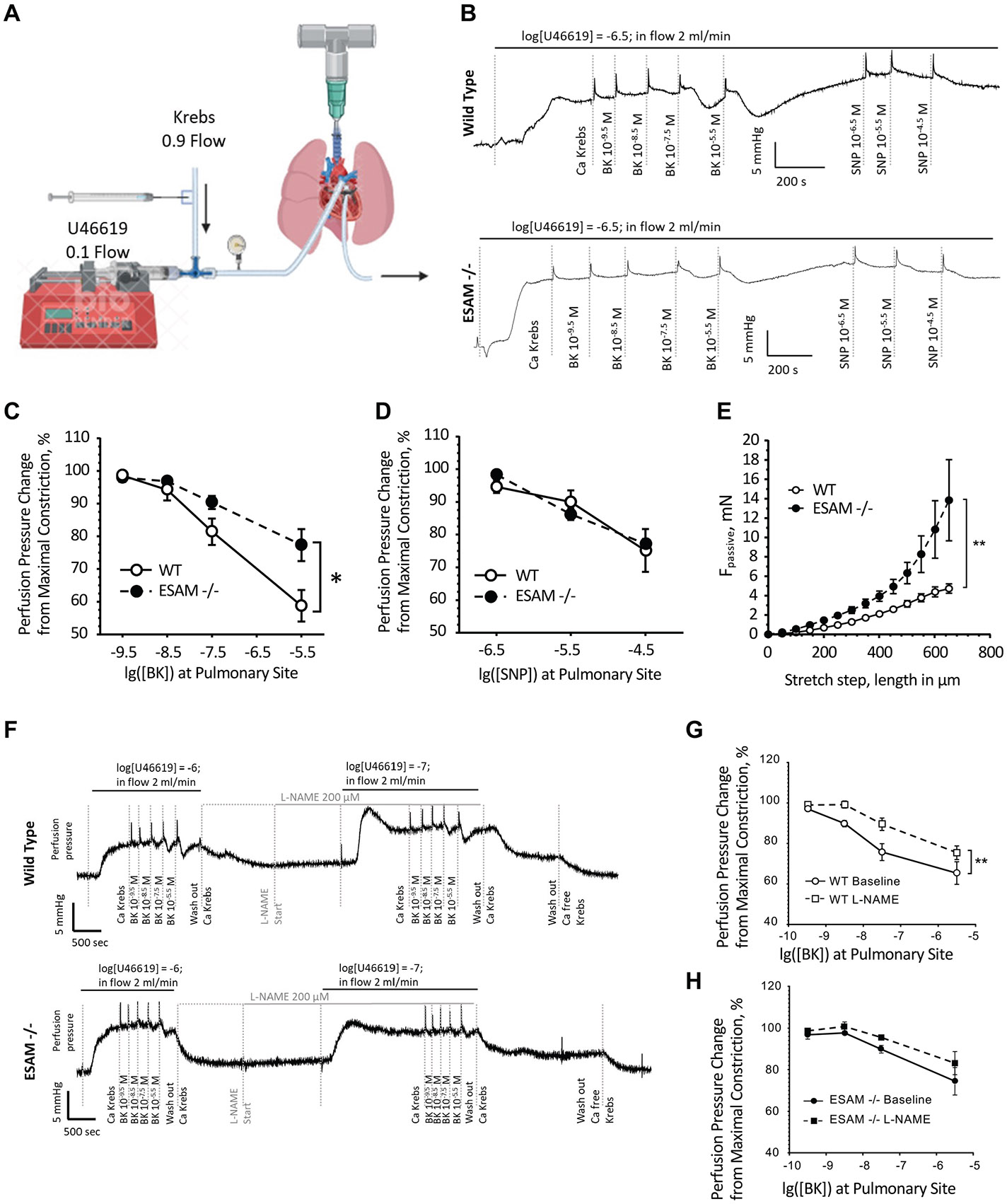
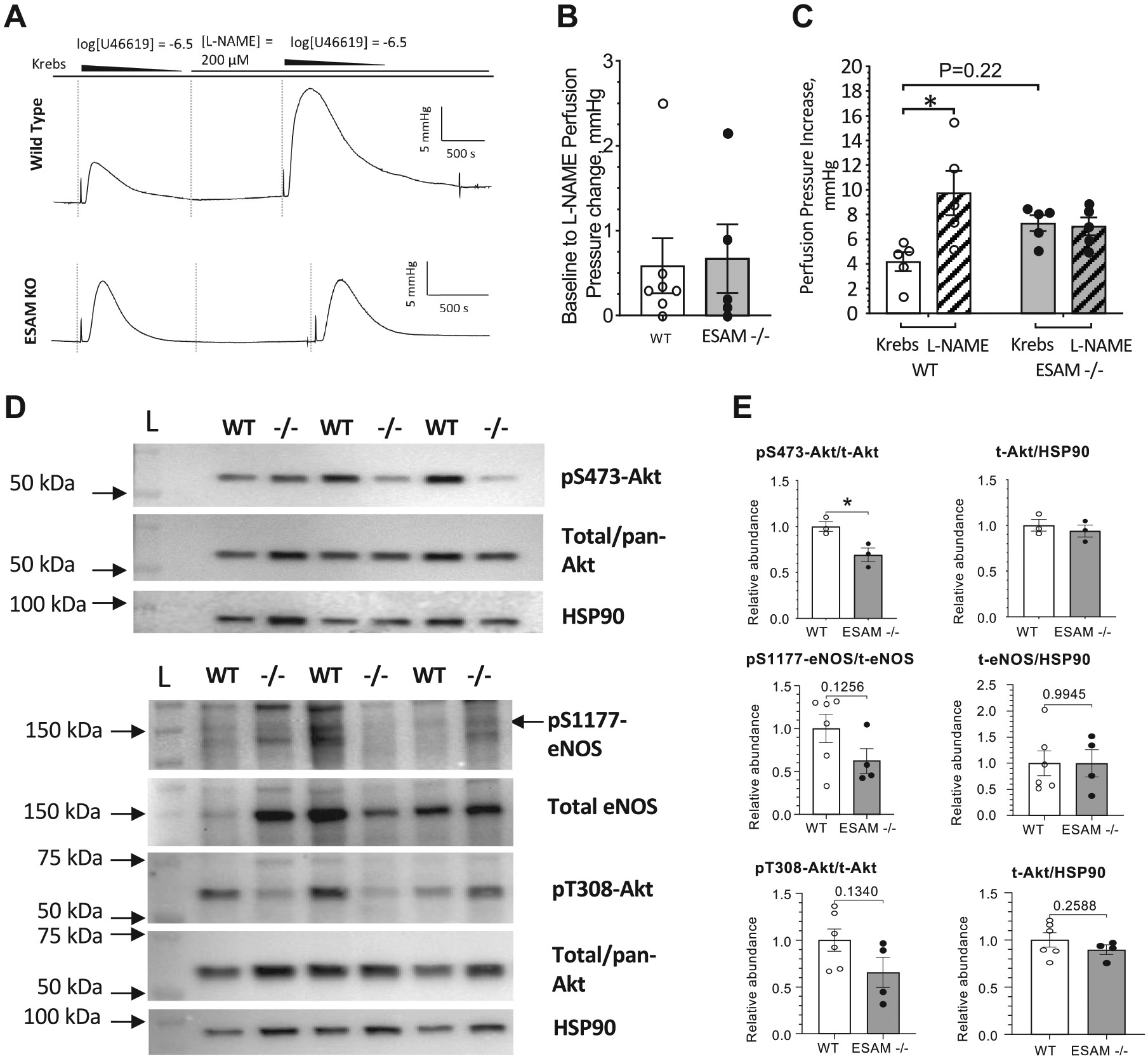
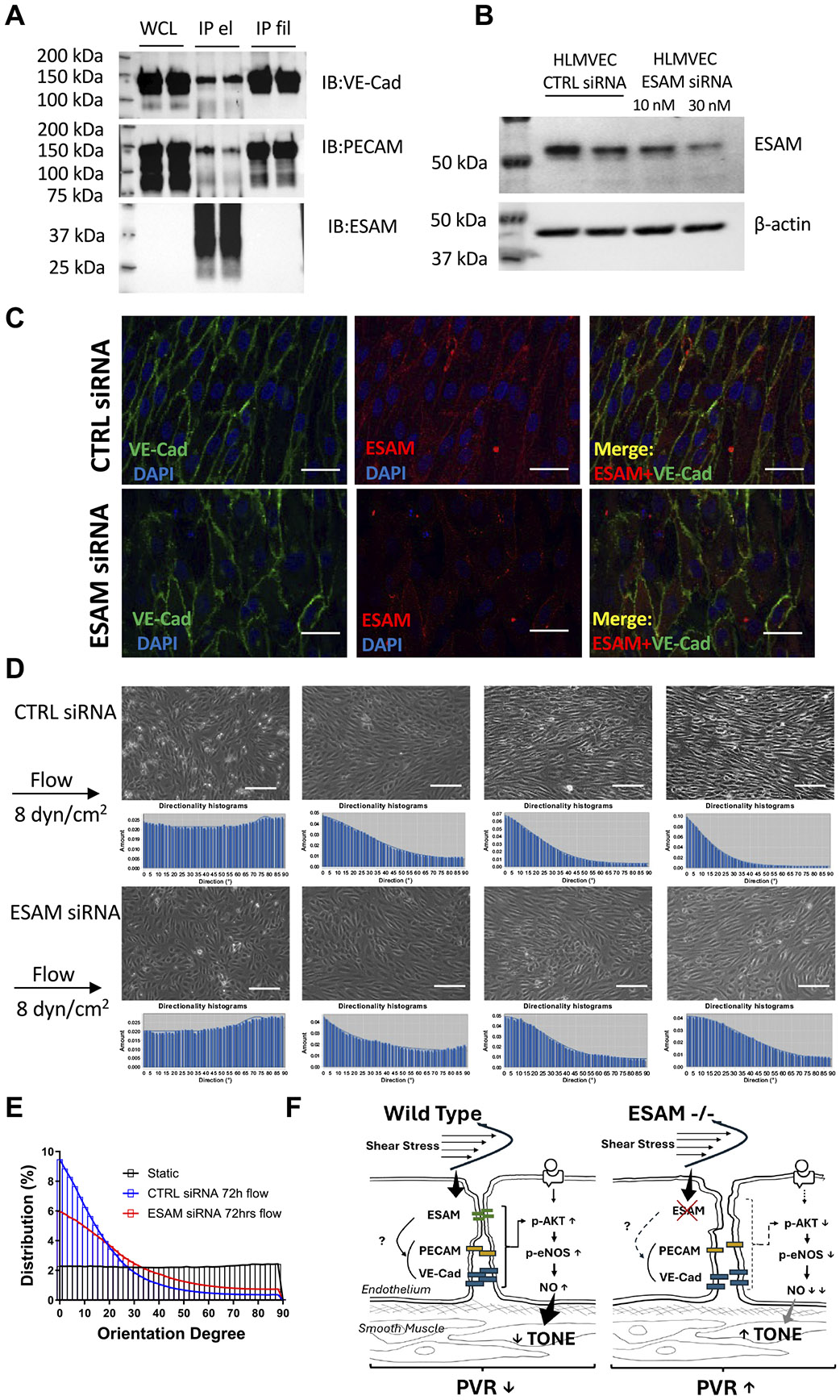
Endothelial cell-selective adhesion molecule (ESAM) is a member of tight junction molecules, highly abundant in the heart and the lung, and plays a role in regulating endothelial cell permeability. We previously reported that mice with genetic ESAM deficiency (ESAM-/-) exhibit coronary microvascular dysfunction leading to the development of left ventricular diastolic dysfunction. Here, we hypothesize that ESAM-/- mice display impairments in the pulmonary vasculature, affecting the overall pulmonary vascular resistance (PVR). We utilized ESAM-/- mice and employed isolated, ventilated, and perfused whole lung preparation to assess PVR independently of cardiac function. PVR was assessed in response to stepwise increases in flow, and also in response to perfusion of the endothelium-dependent agonist, bradykinin, the thromboxane analog, U46619, and the nitric oxide (NO) donor sodium nitroprusside (SNP). We found that PVR, at every applied flow rate, is significantly elevated in ESAM-/- mice compared with WT mice. Bradykinin-induced reduction in PVR and U46619-induced increase in PVR were both diminished in ESAM-/- mice, whereas SNP-induced responses were similar in wild-type (WT) and ESAM-/- mice. Inhibition of NO synthase with N(ω)-nitro-l-arginine methyl ester increased agonist-induced PVR in WT but not in ESAM-/- mice. Pulmonary arteries isolated from ESAM-/- mice exhibited a reduced level of phospho-Ser473-Akt and phospho-Ser1177-eNOS. Furthermore, in human lung microvascular endothelial cells cultured under flow conditions, we found that siRNA-mediated knockdown of ESAM impaired fluid shear stress-induced endothelial cell alignment. Thus, we suggest that ESAM plays an important role in the endothelium-dependent, flow/shear stress- and vasoactive agonist-stimulated, and NO-mediated maintenance of PVR in mice.NEW & NOTEWORTHY Our study reveals a novel role for ESAM in contributing to the maintenance of pulmonary vascular resistance under normal physiological conditions. Employing mice with global genetic deficiency of ESAM and using isolated whole lung preparation, we show significant impairments in nitric oxide-mediated pulmonary artery function. In vitro cell culture studies demonstrate impaired fluid shear stress-induced cell alignment in human lung endothelial cells after siRNA-mediated ESAM knockdown.
Keywords: ESAM; nitric oxide synthase; pulmonary artery; pulmonary vascular resistance.
Copyright © 2025 The Authors.
Conflict of interest statement
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Martìn-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142: 117–127, 1998. doi: 10.1083/jcb.142.1.117. - DOI - PMC - PubMed
-
- Bazzoni G Endothelial tight junctions: permeable barriers of the vessel wall. Thromb Haemost 95: 36–42, 2006. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials