

Targeting T helper 17 cells: emerging strategies for overcoming transplant rejection
- PMID: 39743231
- PMCID: PMC11732763
- DOI: 10.4285/ctr.24.0058
Targeting T helper 17 cells: emerging strategies for overcoming transplant rejection
Abstract
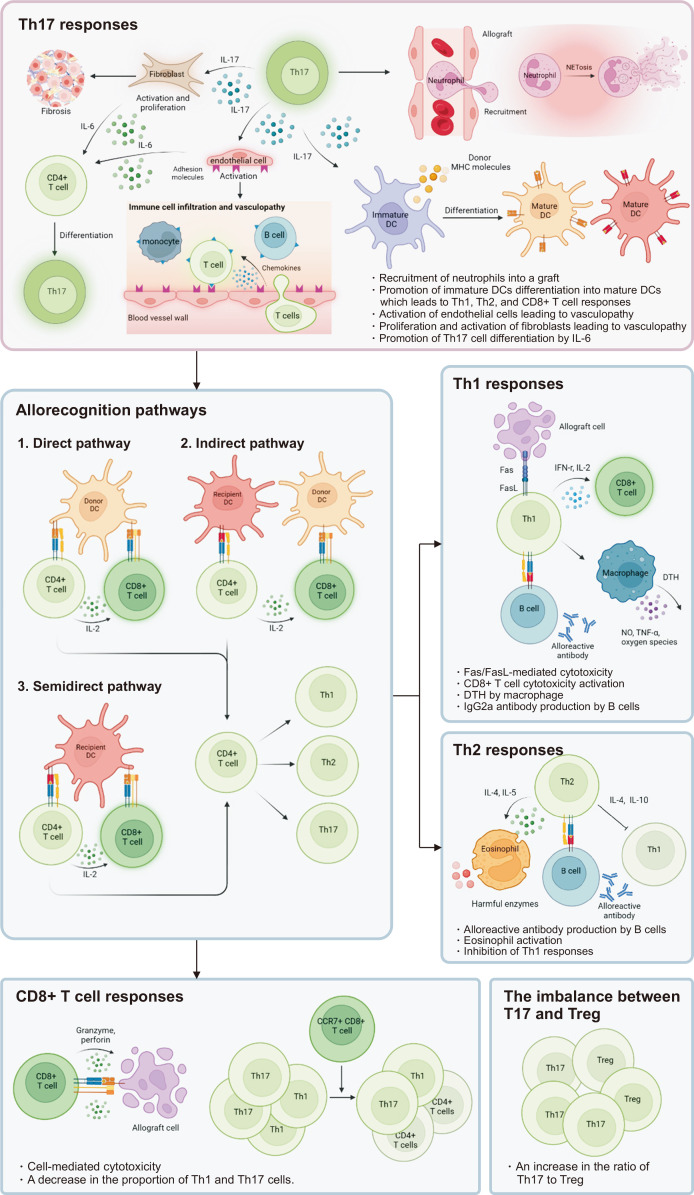
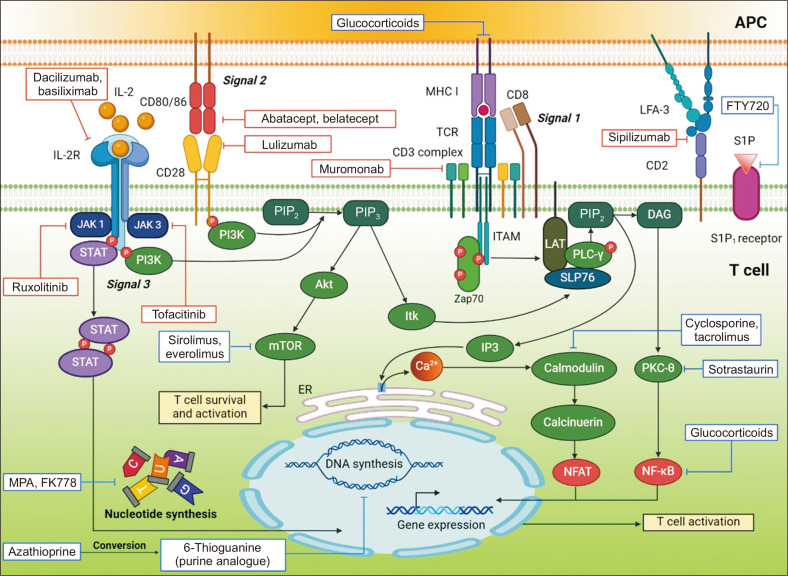
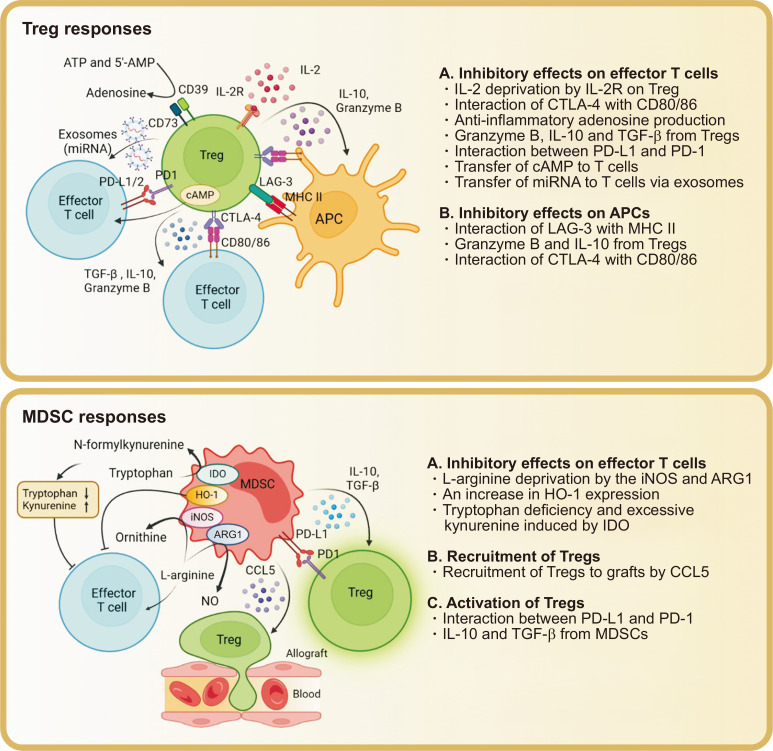
Solid organ transplantation has significantly improved the survival rate of patients with terminal organ failure. However, its success is often compromised by allograft rejection, a process in which T helper 17 (Th17) cells play a crucial role. These cells facilitate rejection by enhancing neutrophil infiltration into the graft and by activating endothelial cells and fibroblasts. Additionally, Th17 cells can trigger the activation of other T cell types, including Th1, Th2, and CD8+ T cells, further contributing to rejection. An imbalance between Th17 and regulatory T cells (Tregs) is known to promote rejection. To counteract this, immunosuppressive drugs have been developed to inhibit T cell activity and foster transplant tolerance. Another approach involves the adoptive transfer of regulatory cells, such as Tregs and myeloid-derived suppressor cells, to dampen T cell functions. This review primarily focuses on the roles of Th17 cells in rejection and their interactions with other T cell subsets. We also explore various strategies aimed at suppressing T cells to induce tolerance.
Keywords: Allograft rejection; Immune tolerance; Th17 cells; Transplantation.
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
