

FBP1 controls liver cancer evolution from senescent MASH hepatocytes
- PMID: 39743585
- PMCID: PMC12168545
- DOI: 10.1038/s41586-024-08317-9
FBP1 controls liver cancer evolution from senescent MASH hepatocytes
Erratum in
-
Author Correction: FBP1 controls liver cancer evolution from senescent MASH hepatocytes.Nature. 2025 Feb;638(8051):E30. doi: 10.1038/s41586-025-08653-4. Nature. 2025. PMID: 39890899 Free PMC article. No abstract available.
Abstract
Hepatocellular carcinoma (HCC) originates from differentiated hepatocytes undergoing compensatory proliferation in livers damaged by viruses or metabolic-dysfunction-associated steatohepatitis (MASH)1. While increasing HCC risk2, MASH triggers p53-dependent hepatocyte senescence3, which we found to parallel hypernutrition-induced DNA breaks. How this tumour-suppressive response is bypassed to license oncogenic mutagenesis and enable HCC evolution was previously unclear. Here we identified the gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) as a p53 target that is elevated in senescent-like MASH hepatocytes but suppressed through promoter hypermethylation and proteasomal degradation in most human HCCs. FBP1 first declines in metabolically stressed premalignant disease-associated hepatocytes and HCC progenitor cells4,5, paralleling the protumorigenic activation of AKT and NRF2. By accelerating FBP1 and p53 degradation, AKT and NRF2 enhance the proliferation and metabolic activity of previously senescent HCC progenitors. The senescence-reversing and proliferation-supportive NRF2-FBP1-AKT-p53 metabolic switch, operative in mice and humans, also enhances the accumulation of DNA-damage-induced somatic mutations needed for MASH-to-HCC progression.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: M.K. is a founder and stockholder in Elgia Pharmaceuticals and received research support from Merck and Janssen Pharmaceuticals and holds a patent for the use of MUP-uPA mice as a NASH-HCC model. L.B.A. is a compensated consultant and has equity interest in io9 and Genome Insight; his spouse is an employee of Biotheranostics. L.B.A. is also listed as an inventor on US Patent 10,776,718 for source identification by non-negative matrix factorization. L.B.A. declares US provisional applications with the following serial numbers: 63/289,601, 63/269,033, 63/483,237, 63/366,392 and 63/367,846. L.B.A. and S.P.N. also declare US provisional applications with serial numbers 63/412,835 and 63/492,348. The other authors declare no competing interests.
Figures






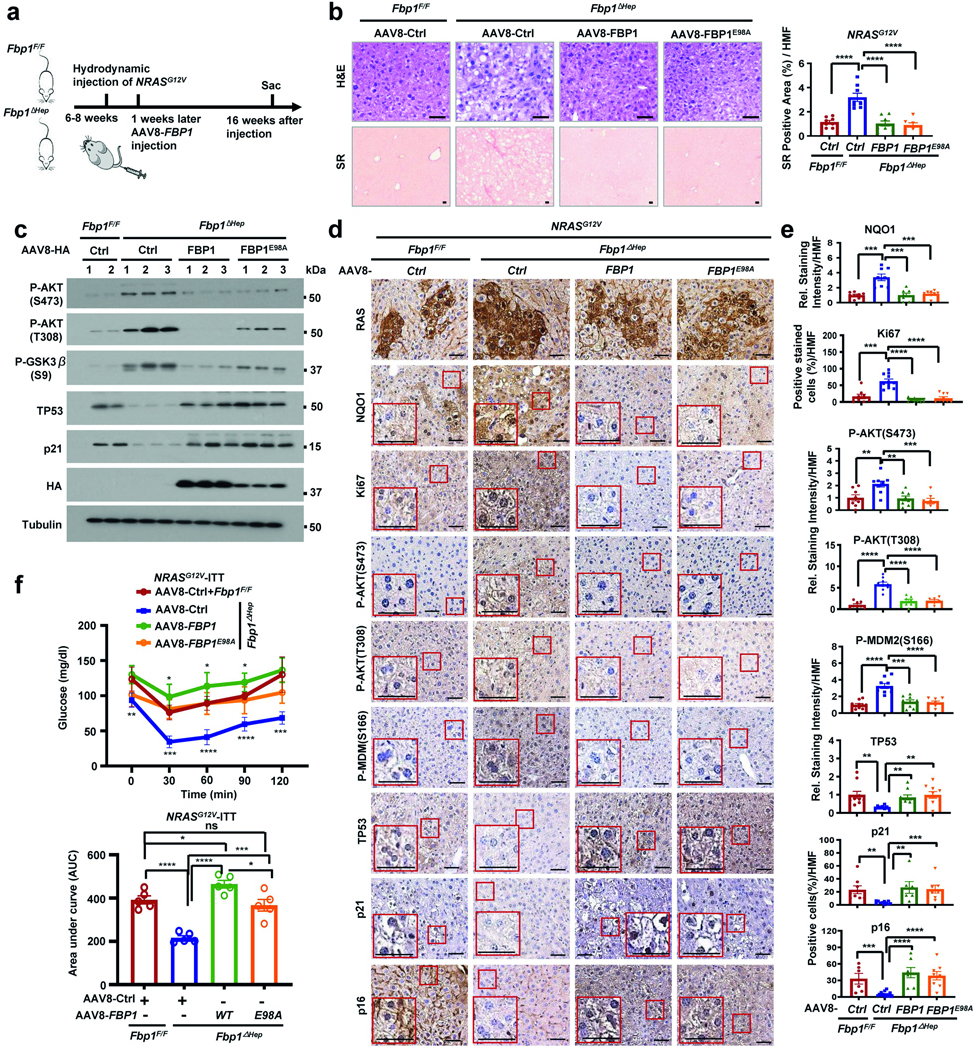
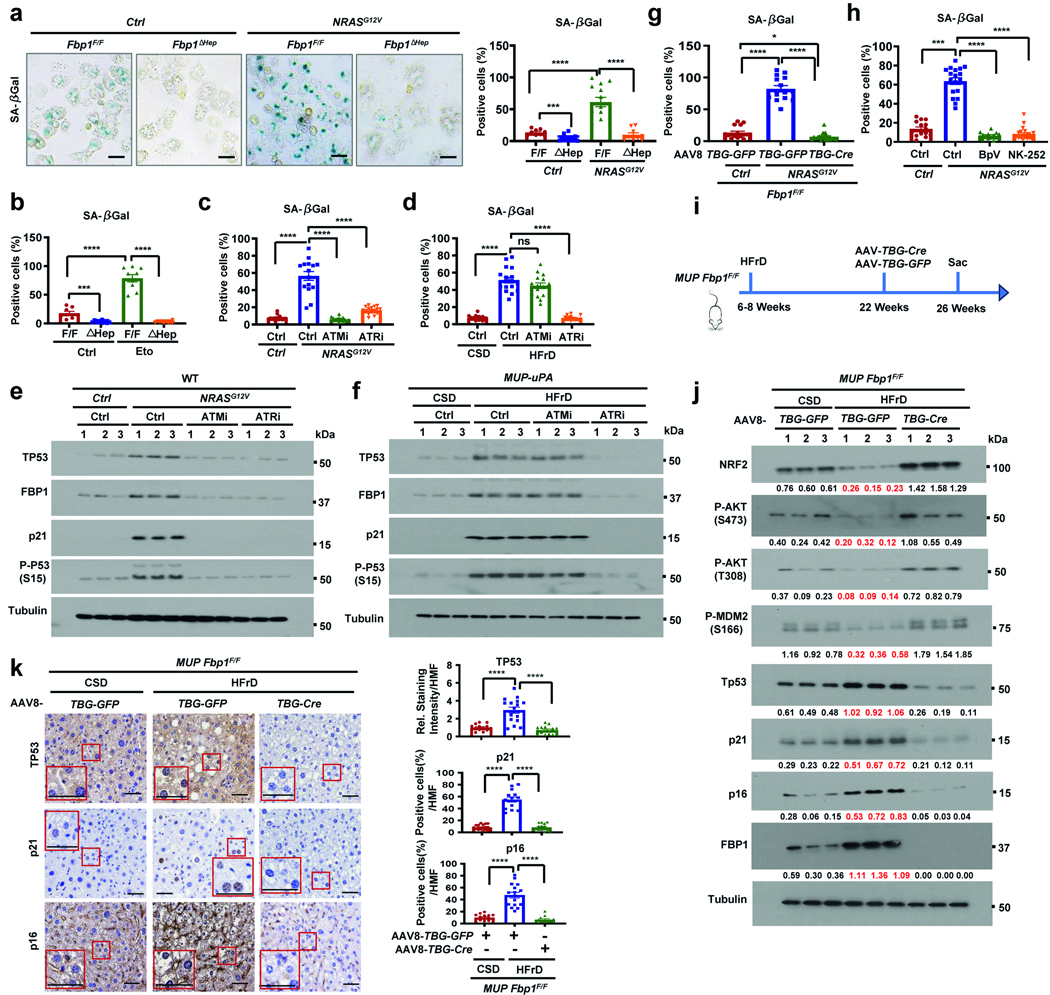




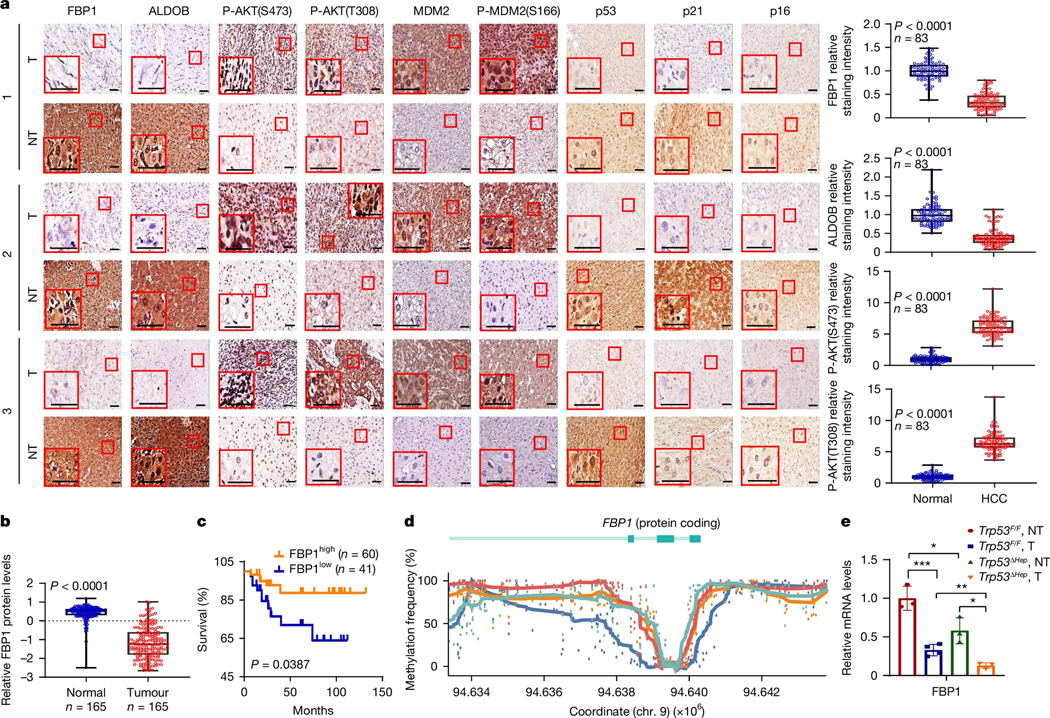





References
-
- Maeda S, Kamata H, Luo JL, Leffert H & Karin M IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121, 977–990 (2005). - PubMed
-
- Ferreira-Gonzalez S, Rodrigo-Torres D, Gadd VL & Forbes SJ Cellular senescence in liver disease and regeneration. Semin. Liver Dis 41, 50–66 (2021). - PubMed
MeSH terms
Substances
Grants and funding
- R01 DK120714/DK/NIDDK NIH HHS/United States
- R35 CA220483/CA/NCI NIH HHS/United States
- S10 OD032398/OD/NIH HHS/United States
- P30 DK120515/DK/NIDDK NIH HHS/United States
- P01 AG073084/AG/NIA NIH HHS/United States
- KL2 TR001444/TR/NCATS NIH HHS/United States
- R01 DK133930/DK/NIDDK NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- R01 CA281784/CA/NCI NIH HHS/United States
- P01 CA281819/CA/NCI NIH HHS/United States
- R01 CA269919/CA/NCI NIH HHS/United States
- R01 CA234128/CA/NCI NIH HHS/United States
- R01 DK133448/DK/NIDDK NIH HHS/United States
- R01 ES030993/ES/NIEHS NIH HHS/United States
- R01 ES032547/ES/NIEHS NIH HHS/United States
- R01 DK137061/DK/NIDDK NIH HHS/United States
- R01 DK099205/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
