

Gliomagenesis mimics an injury response orchestrated by neural crest-like cells
- PMID: 39743595
- PMCID: PMC11821533
- DOI: 10.1038/s41586-024-08356-2
Gliomagenesis mimics an injury response orchestrated by neural crest-like cells
Abstract
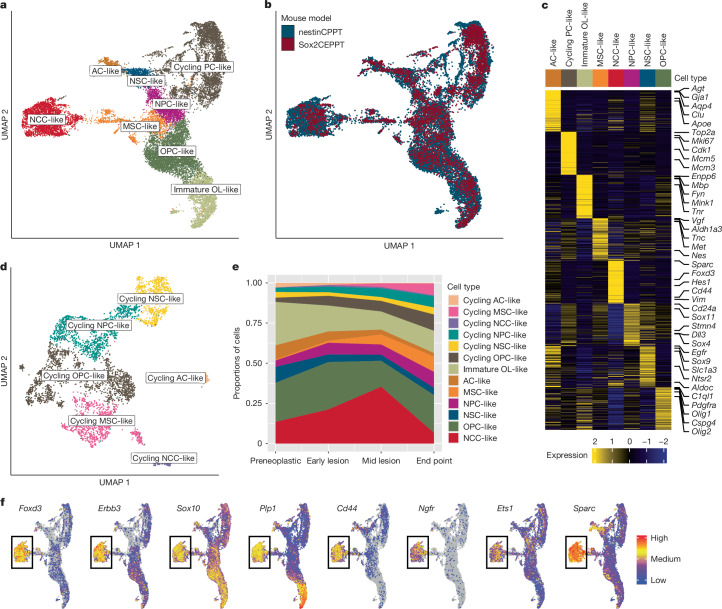
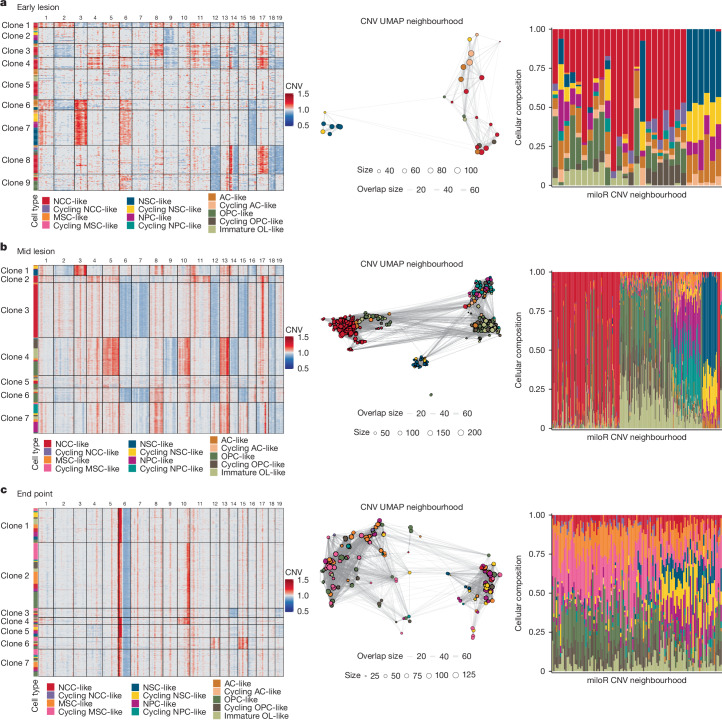
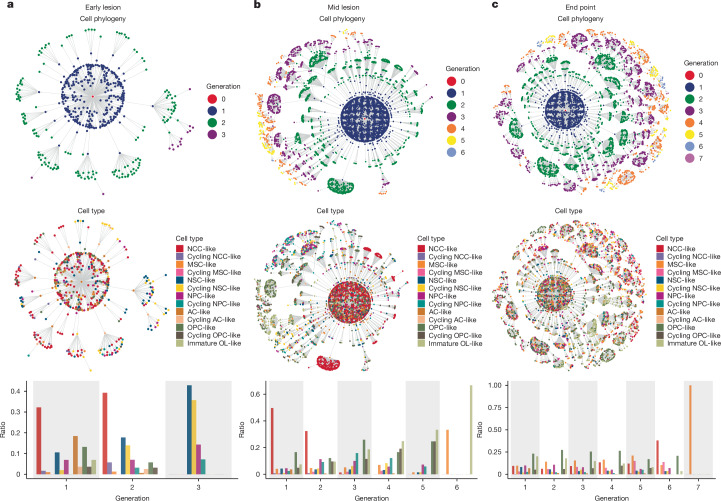
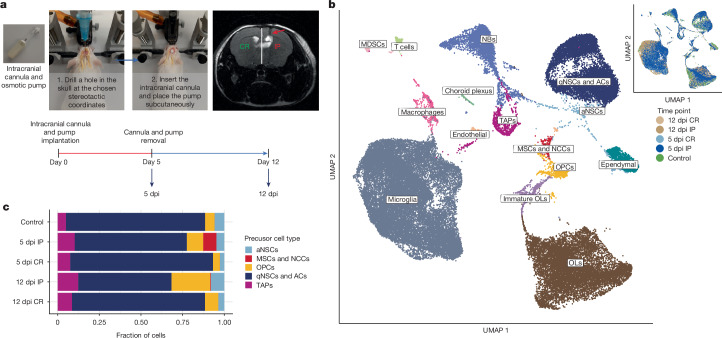
Glioblastoma is an incurable brain malignancy. By the time of clinical diagnosis, these tumours exhibit a degree of genetic and cellular heterogeneity that provides few clues to the mechanisms that initiate and drive gliomagenesis1,2. Here, to explore the early steps in gliomagenesis, we utilized conditional gene deletion and lineage tracing in tumour mouse models, coupled with serial magnetic resonance imaging, to initiate and then closely track tumour formation. We isolated labelled and unlabelled cells at multiple stages-before the first visible abnormality, at the time of the first visible lesion, and then through the stages of tumour growth-and subjected cells of each stage to single-cell profiling. We identify a malignant cell state with a neural crest-like gene expression signature that is highly abundant in the early stages, but relatively diminished in the late stage of tumour growth. Genomic analysis based on the presence of copy number alterations suggests that these neural crest-like states exist as part of a heterogeneous clonal hierarchy that evolves with tumour growth. By exploring the injury response in wounded normal mouse brains, we identify cells with a similar signature that emerge following injury and then disappear over time, suggesting that activation of an injury response program occurs during tumorigenesis. Indeed, our experiments reveal a non-malignant injury-like microenvironment that is initiated in the brain following oncogene activation in cerebral precursor cells. Collectively, our findings provide insight into the early stages of glioblastoma, identifying a unique cell state and an injury response program tied to early tumour formation. These findings have implications for glioblastoma therapies and raise new possibilities for early diagnosis and prevention of disease.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
-
- Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature432, 396–401 (2004). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
