

High-resolution genomic history of early medieval Europe
- PMID: 39743601
- PMCID: PMC11693606
- DOI: 10.1038/s41586-024-08275-2
High-resolution genomic history of early medieval Europe
Abstract
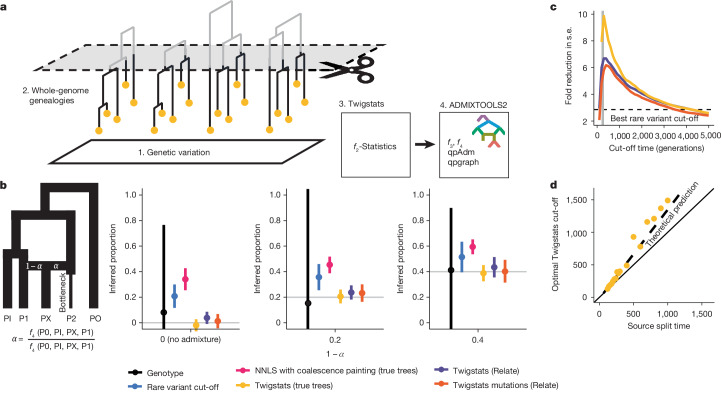
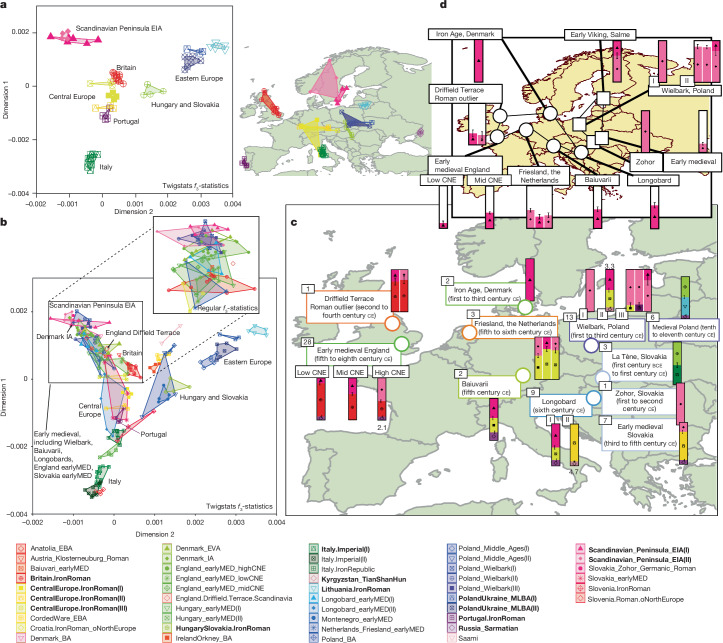
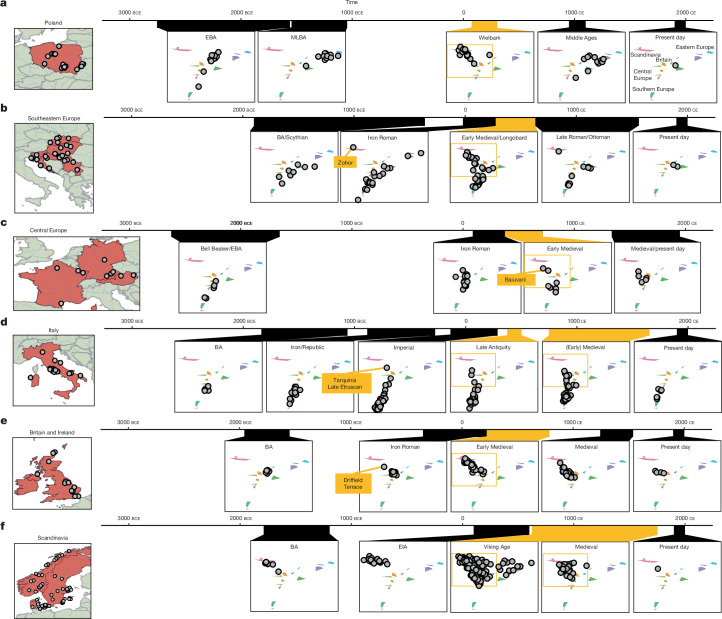
Many known and unknown historical events have remained below detection thresholds of genetic studies because subtle ancestry changes are challenging to reconstruct. Methods based on shared haplotypes1,2 and rare variants3,4 improve power but are not explicitly temporal and have not been possible to adopt in unbiased ancestry models. Here we develop Twigstats, an approach of time-stratified ancestry analysis that can improve statistical power by an order of magnitude by focusing on coalescences in recent times, while remaining unbiased by population-specific drift. We apply this framework to 1,556 available ancient whole genomes from Europe in the historical period. We are able to model individual-level ancestry using preceding genomes to provide high resolution. During the first half of the first millennium CE, we observe at least two different streams of Scandinavian-related ancestry expanding across western, central and eastern Europe. By contrast, during the second half of the first millennium CE, ancestry patterns suggest the regional disappearance or substantial admixture of these ancestries. In Scandinavia, we document a major ancestry influx by approximately 800 CE, when a large proportion of Viking Age individuals carried ancestry from groups related to central Europe not seen in individuals from the early Iron Age. Our findings suggest that time-stratified ancestry analysis can provide a higher-resolution lens for genetic history.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures











References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
