

Progressive lung fibrosis: reprogramming a genetically vulnerable bronchoalveolar epithelium
- PMID: 39744946
- PMCID: PMC11684817
- DOI: 10.1172/JCI183836
Progressive lung fibrosis: reprogramming a genetically vulnerable bronchoalveolar epithelium
Abstract
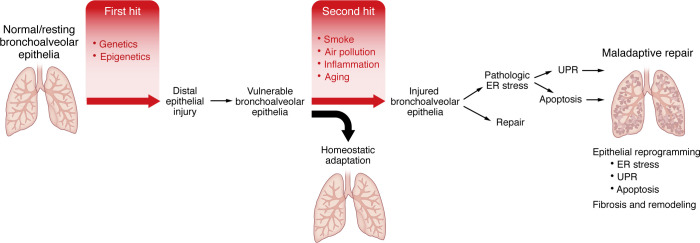
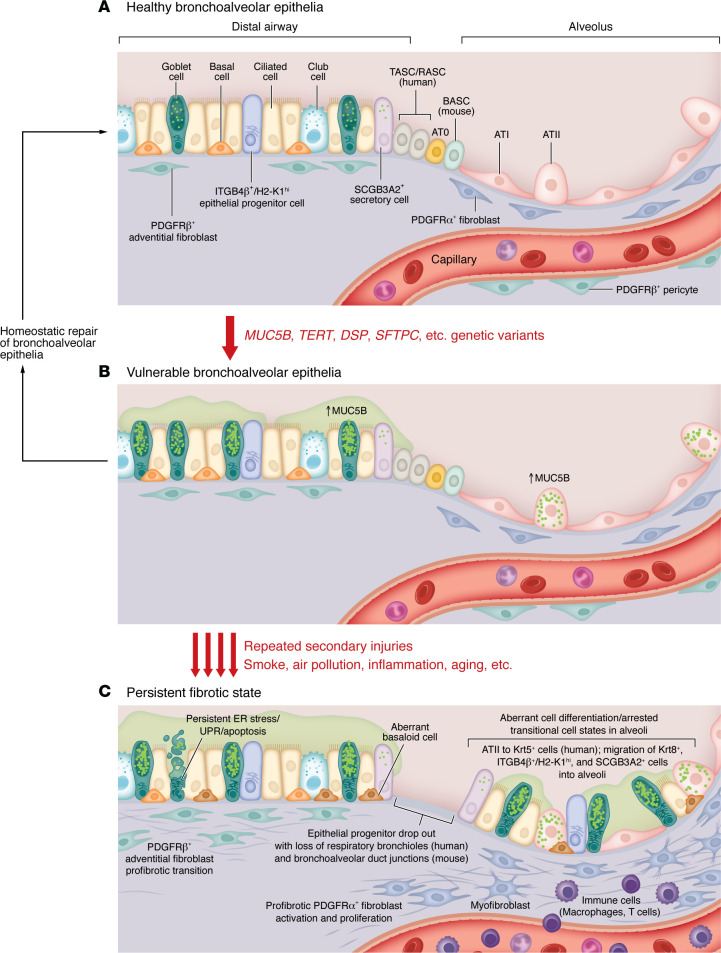
Idiopathic pulmonary fibrosis (IPF) is etiologically complex, with well-documented genetic and nongenetic origins. In this Review, we speculate that the development of IPF requires two hits: the first establishes a vulnerable bronchoalveolar epithelium, and the second triggers mechanisms that reprogram distal epithelia to initiate and perpetuate a profibrotic phenotype. While vulnerability of the bronchoalveolar epithelia is most often driven by common or rare genetic variants, subsequent injury of the bronchoalveolar epithelia results in persistent changes in cell biology that disrupt tissue homeostasis and activate fibroblasts. The dynamic biology of IPF can best be contextualized etiologically and temporally, including stages of vulnerability, early disease, and persistent and progressive lung fibrosis. These dimensions of IPF highlight critical mechanisms that adversely disrupt epithelial function, activate fibroblasts, and lead to lung remodeling. Together with better recognition of early disease, this conceptual approach should lead to the development of novel therapeutics directed at the etiologic and temporal drivers of lung fibrosis that will ultimately transform the care of patients with IPF from palliative to curative.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
