

Decomposition of the pangenome matrix reveals a structure in gene distribution in the Escherichia coli species
- PMID: 39745367
- PMCID: PMC11774025
- DOI: 10.1128/msphere.00532-24
Decomposition of the pangenome matrix reveals a structure in gene distribution in the Escherichia coli species
Abstract
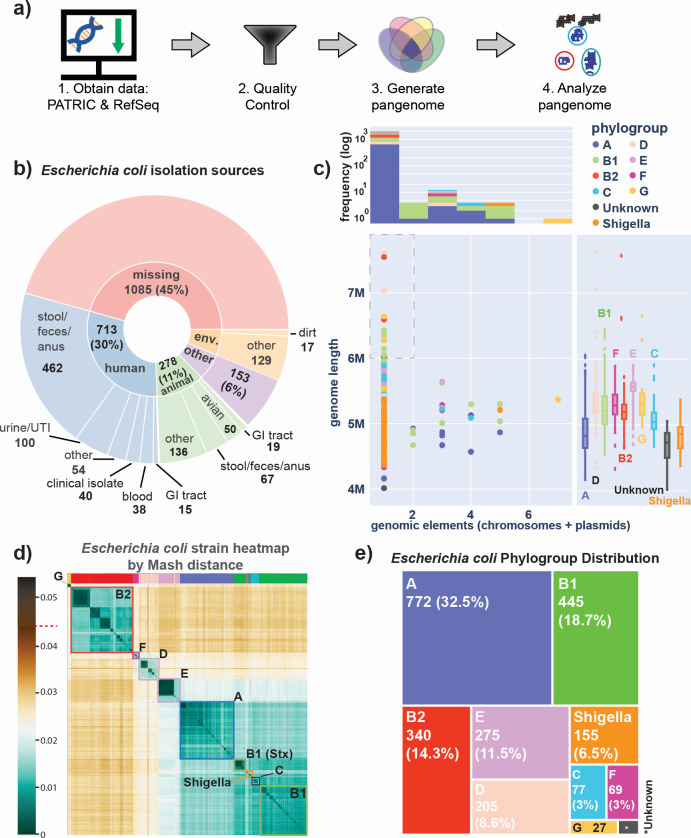
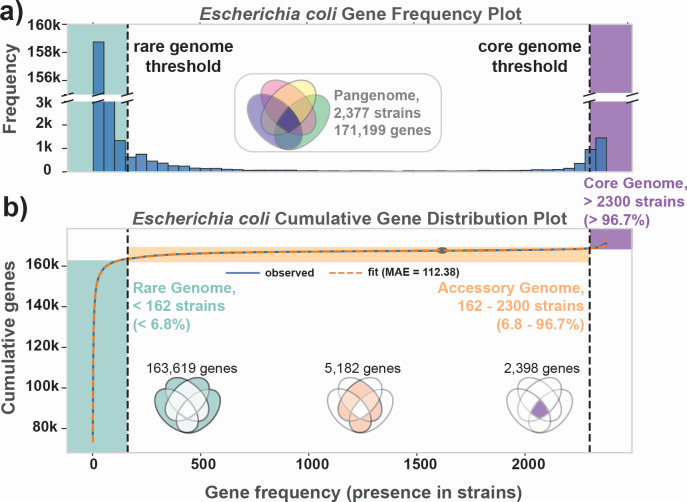
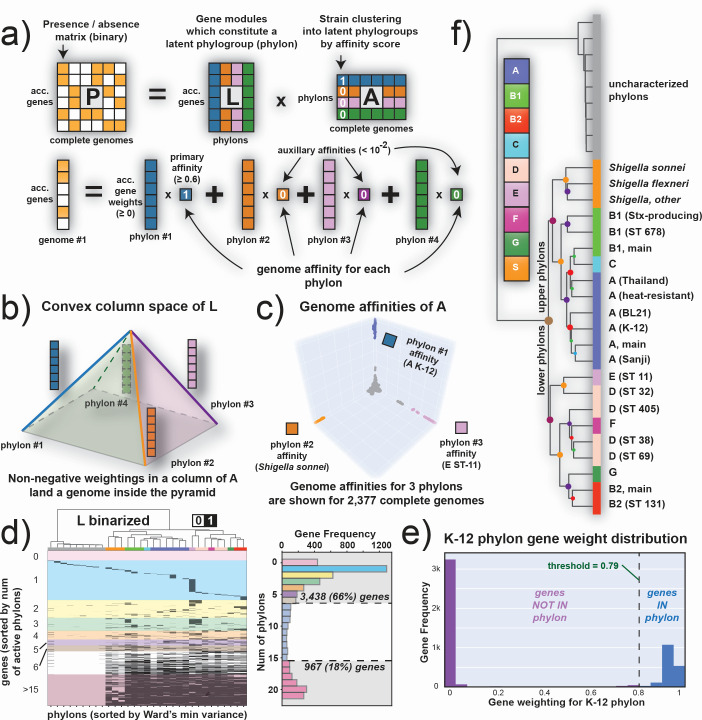
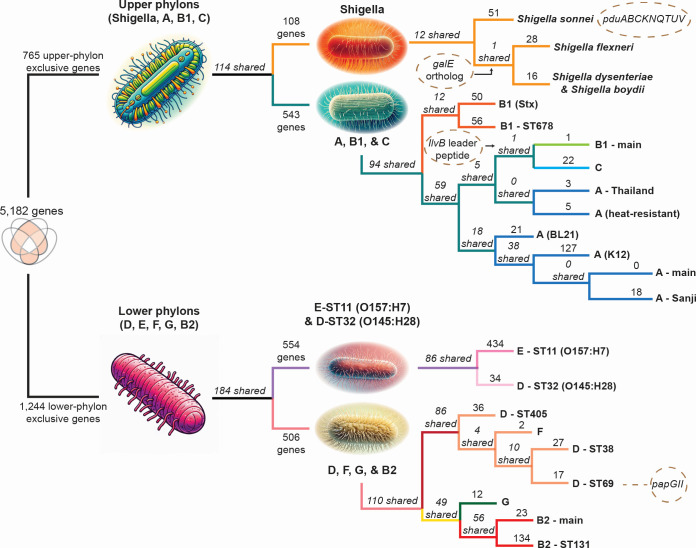
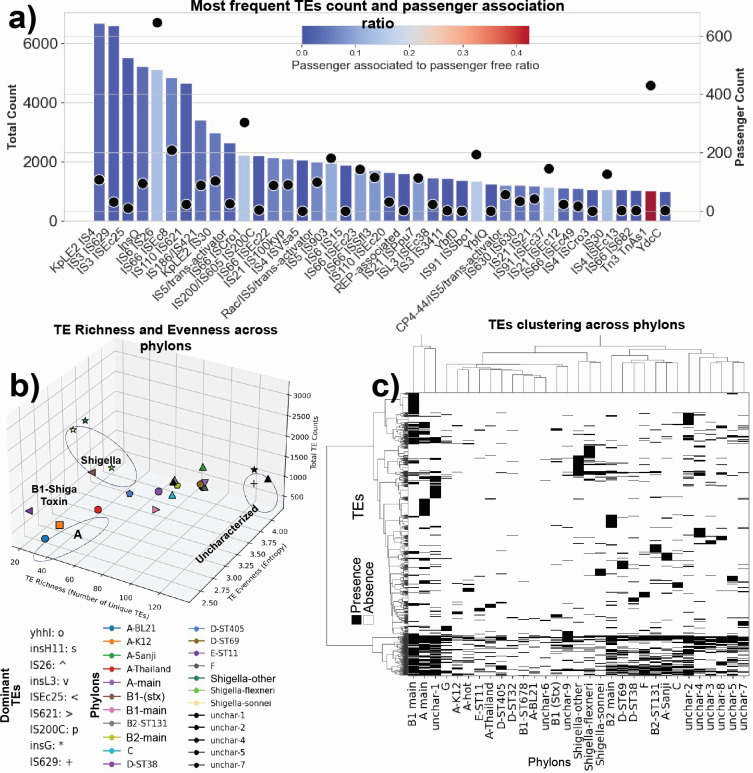
Thousands of complete genome sequences for strains of a species that are now available enable the advancement of pangenome analytics to a new level of sophistication. We collected 2,377 publicly available complete genomes of Escherichia coli for detailed pangenome analysis. The core genome and accessory genomes consisted of 2,398 and 5,182 genes, respectively. We developed a machine learning approach to define the accessory genes characterizing the major phylogroups of E. coli plus Shigella: A, B1, B2, C, D, E, F, G, and Shigella. The analysis resulted in a detailed structure of the genetic basis of the phylogroups' differential traits. This pangenome structure was largely consistent with a housekeeping-gene-based MLST distribution, sequence-based Mash distance, and the Clermont quadruplex classification. The rare genome (consisting of genes found in <6.8% of all strains) consisted of 163,619 genes, about 79% of which represented variations of 315 underlying transposon elements. This analysis generated a mathematical definition of the genetic basis for a species.
Importance: The comprehensive analysis of the pangenome of Escherichia coli presented in this study marks a significant advancement in understanding bacterial genetic diversity. By employing machine learning techniques to analyze 2,377 complete E. coli genomes, the study provides a detailed mapping of core, accessory, and rare genes. This approach reveals the genetic basis for differential traits across phylogroups, offering insights into pathogenicity, antibiotic resistance, and evolutionary adaptations. The findings enhance the potential for genome-based diagnostics and pave the way for future studies aimed at achieving a global genetic definition of bacterial phylogeny.
Keywords: Escherichia coli; Shigella; computational biology; genome analysis; genomics; typing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453 - DOI - PubMed
-
- Kris A. Wetterstrand MS. 2019. The cost of sequencing a human genome. NHGRI. Available from: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genom.... Retrieved 18 Apr 2023.
-
- Olson RD, Assaf R, Brettin T, Conrad N, Cucinell C, Davis JJ, Dempsey DM, Dickerman A, Dietrich EM, Kenyon RW, et al. 2023. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res 51:D678–D689. doi: 10.1093/nar/gkac1003 - DOI - PMC - PubMed
