

Identification and characterization of Faecalibacterium prophages rich in diversity-generating retroelements
- PMID: 39745426
- PMCID: PMC11792537
- DOI: 10.1128/spectrum.01066-24
Identification and characterization of Faecalibacterium prophages rich in diversity-generating retroelements
Abstract
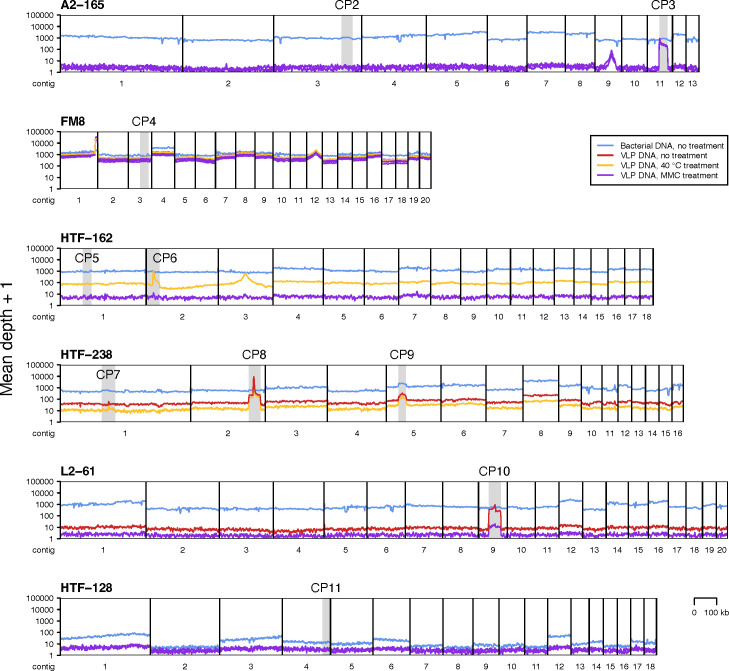
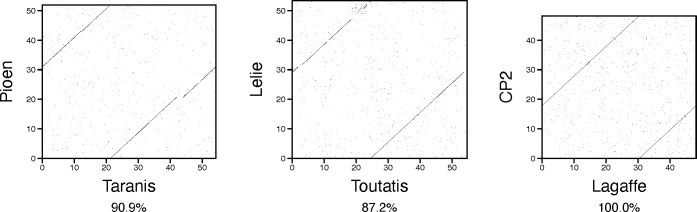
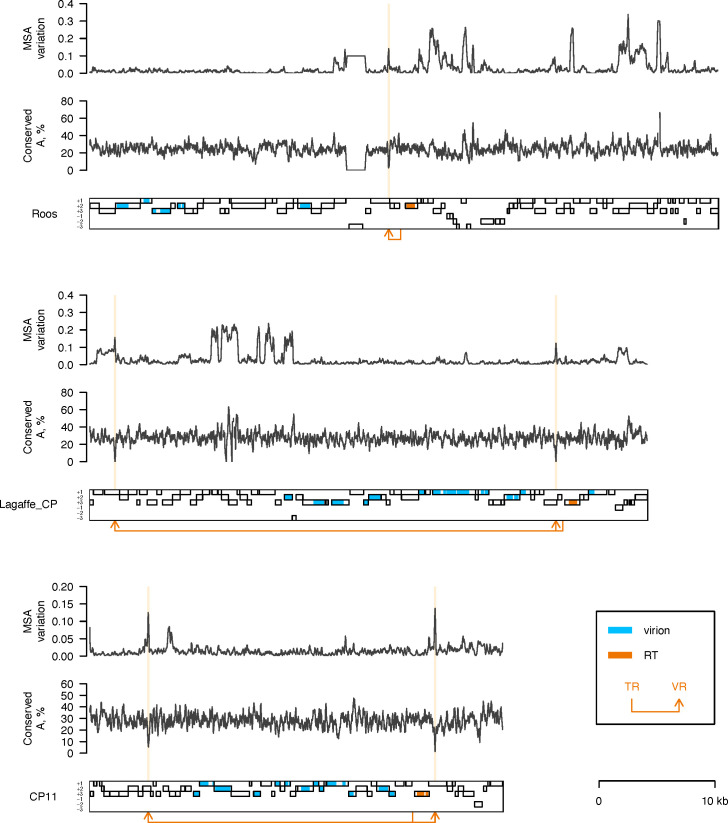
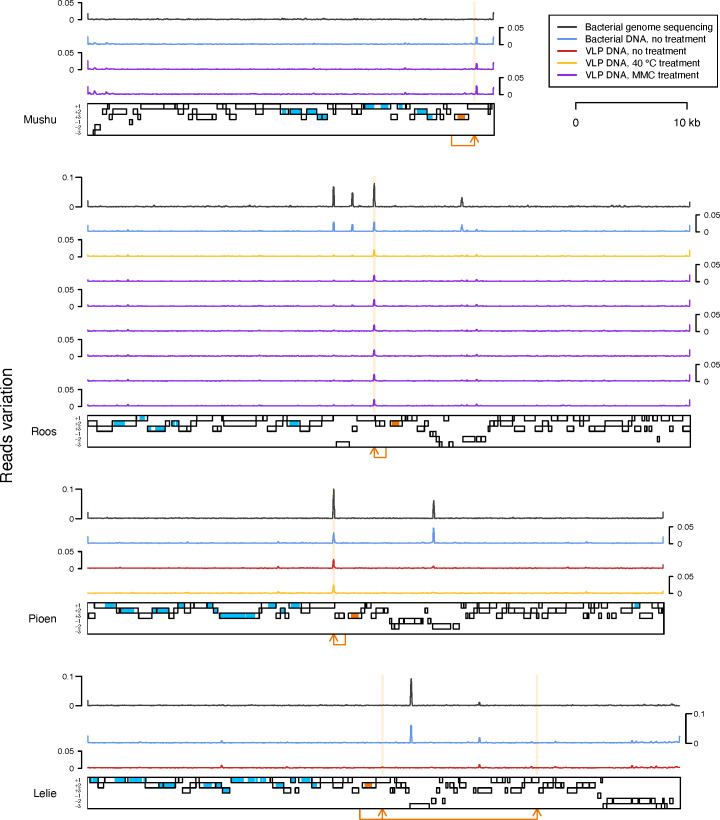
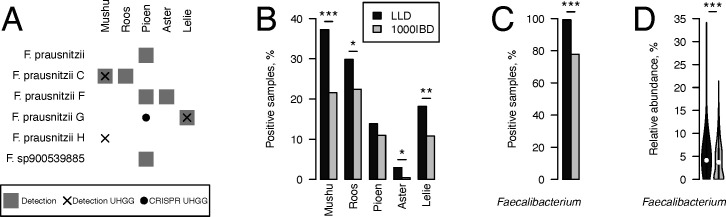
Metagenomics has revealed the incredible diversity of phages within the human gut. However, very few of these phages have been subjected to in-depth experimental characterization. One promising method of obtaining novel phages for experimental characterization is through induction of the prophages integrated into the genomes of cultured gut bacteria. Here, we developed a bioinformatic approach to prophage identification that builds on prophage genomic properties, existing prophage-detecting software, and publicly available virome sequencing data. We applied our approach to 22 strains of bacteria belonging to the genus Faecalibacterium, resulting in identification of 15 candidate prophages, and validated the approach by demonstrating the activity of five prophages from four of the strains. The genomes of three active phages were identical or similar to those of known phages, while the other two active phages were not represented in the Viral RefSeq database. Four of the active phages possessed a diversity-generating retroelement (DGR), and one retroelement had two variable regions. DGRs of two phages were active at the time of the induction experiments, as evidenced by nucleotide variation in sequencing reads. We also predicted that the host range of two active phages may include multiple bacterial species. Finally, we noted that four phages were less prevalent in the metagenomes of inflammatory bowel disease patients compared to a general population cohort, a difference mainly explained by differences in the abundance of the host bacteria. Our study highlights the utility of prophage identification and induction for unraveling phage molecular mechanisms and ecological interactions.IMPORTANCEWhile hundreds of thousands of phage genomes have been discovered in metagenomics studies, only a few of these phages have been characterized experimentally. Here, we explore phage characterization through bioinformatic identification of prophages in genomes of cultured bacteria, followed by prophage induction. Using this approach, we detect the activity of five prophages in four strains of commensal gut bacteria Faecalibacterium. We further note that four of the prophages possess diversity-generating retroelements implicated in rapid mutation of phage genome loci associated with phage-host and phage-environment interactions and analyze the intricate patterns of retroelement activity. Our study highlights the potential of prophage characterization for elucidating complex molecular mechanisms employed by the phages.
Keywords: DGR; bacteriophage; microbiome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
A diversity-generating retroelement encoded by a globally ubiquitous Bacteroides phage.Microbiome. 2018 Oct 23;6(1):191. doi: 10.1186/s40168-018-0573-6. Microbiome. 2018. PMID: 30352623 Free PMC article.
-
Host population structure and species resolution reveal prophage transmission dynamics.mBio. 2024 Oct 16;15(10):e0237724. doi: 10.1128/mbio.02377-24. Epub 2024 Sep 24. mBio. 2024. PMID: 39315801 Free PMC article.
-
Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes.Microbiome. 2018 Apr 3;6(1):65. doi: 10.1186/s40168-018-0452-1. Microbiome. 2018. PMID: 29615108 Free PMC article.
-
Phage hunters: Computational strategies for finding phages in large-scale 'omics datasets.Virus Res. 2018 Jan 15;244:110-115. doi: 10.1016/j.virusres.2017.10.019. Epub 2017 Nov 1. Virus Res. 2018. PMID: 29100906 Review.
-
Impact of prophages on gut microbiota and disease associations.Microb Pathog. 2025 Jul;204:107642. doi: 10.1016/j.micpath.2025.107642. Epub 2025 Apr 27. Microb Pathog. 2025. PMID: 40300731 Review.
Cited by
-
The colonic mucosal virome in inflammatory bowel disease reveals Crassvirales depletion and disease-specific virome features.Gut Microbes. 2025 Dec;17(1):2539450. doi: 10.1080/19490976.2025.2539450. Epub 2025 Aug 3. Gut Microbes. 2025. PMID: 40754936 Free PMC article.
-
The Microbiome and Metabolic Dysfunction-Associated Steatotic Liver Disease.Int J Mol Sci. 2025 Mar 22;26(7):2882. doi: 10.3390/ijms26072882. Int J Mol Sci. 2025. PMID: 40243472 Free PMC article. Review.
-
Single cell viral tagging of Faecalibacterium prausnitzii reveals rare bacteriophages omitted by other techniques.Gut Microbes. 2025 Dec;17(1):2526719. doi: 10.1080/19490976.2025.2526719. Epub 2025 Aug 3. Gut Microbes. 2025. PMID: 40754853 Free PMC article.
References
-
- Camargo AP, Nayfach S, Chen I-MA, Palaniappan K, Ratner A, Chu K, Ritter SJ, Reddy TBK, Mukherjee S, Schulz F, Call L, Neches RY, Woyke T, Ivanova NN, Eloe-Fadrosh EA, Kyrpides NC, Roux S. 2023. IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res 51:D733–D743. doi:10.1093/nar/gkac1037 - DOI - PMC - PubMed
-
- Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, Felts B, Dinsdale EA, Mokili JL, Edwards RA. 2014. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 5:4498. doi:10.1038/ncomms5498 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
