

In defence of ferroptosis
- PMID: 39746918
- PMCID: PMC11696223
- DOI: 10.1038/s41392-024-02088-5
In defence of ferroptosis
Abstract
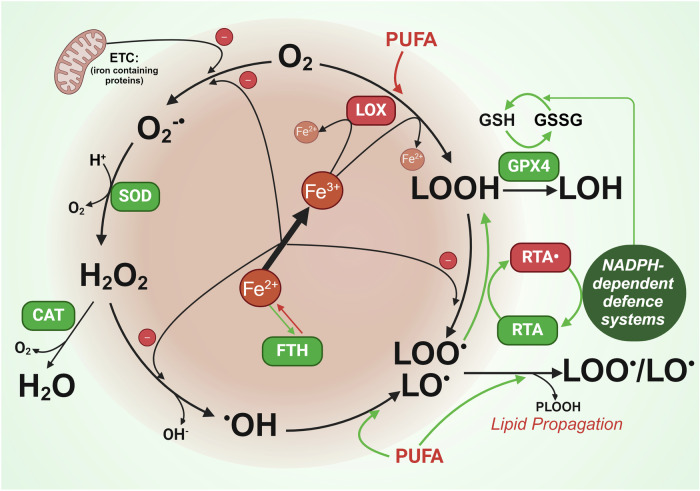
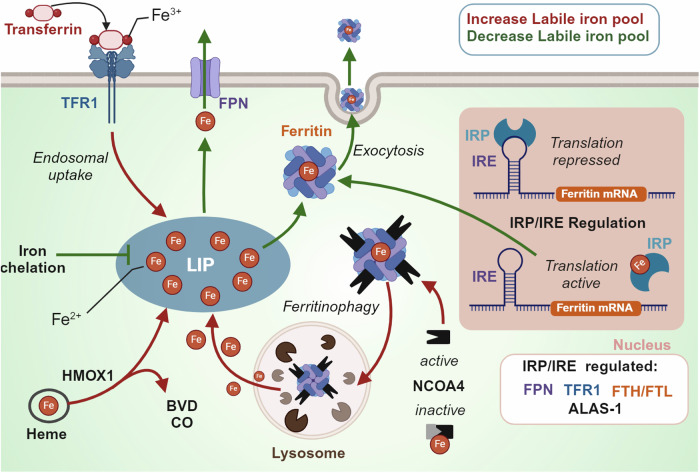
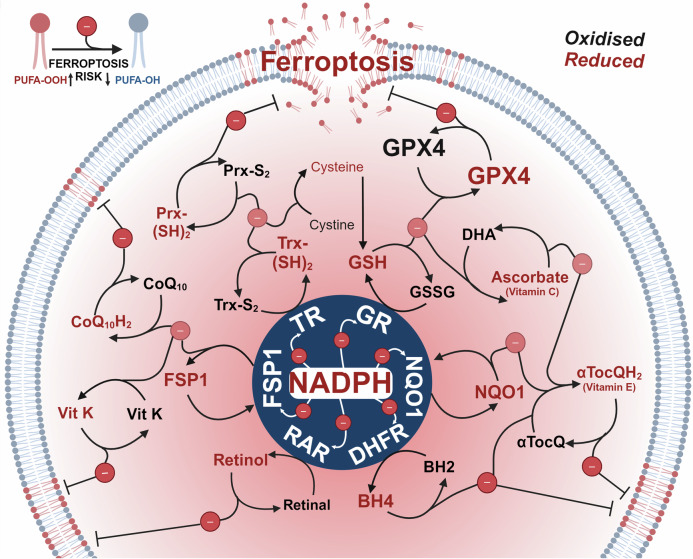
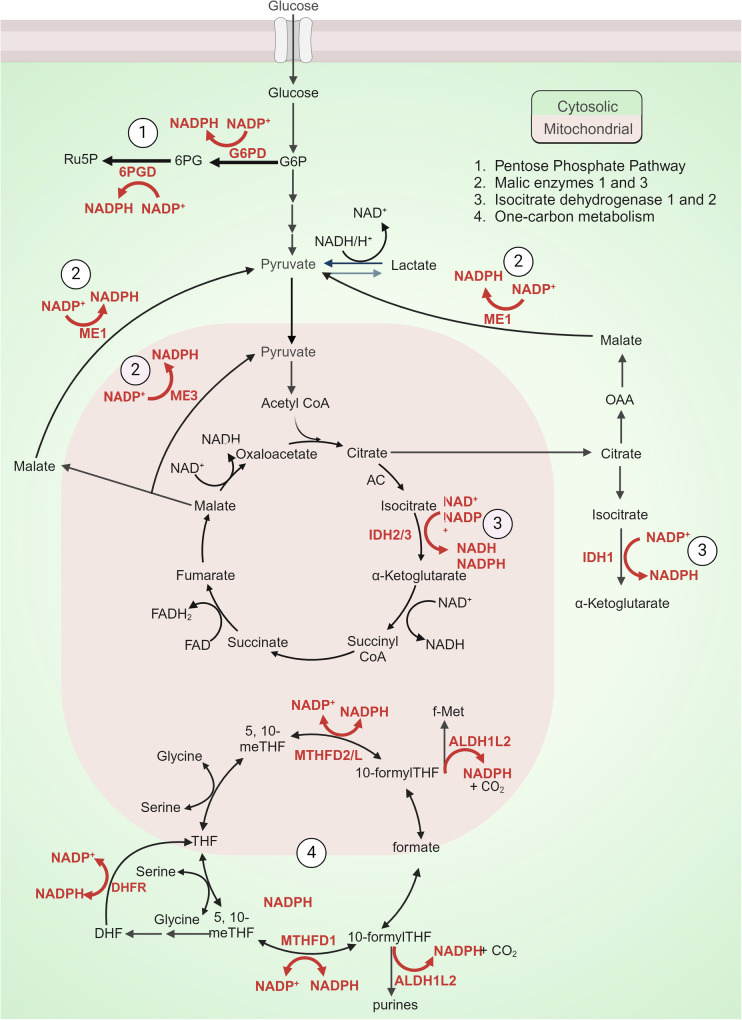
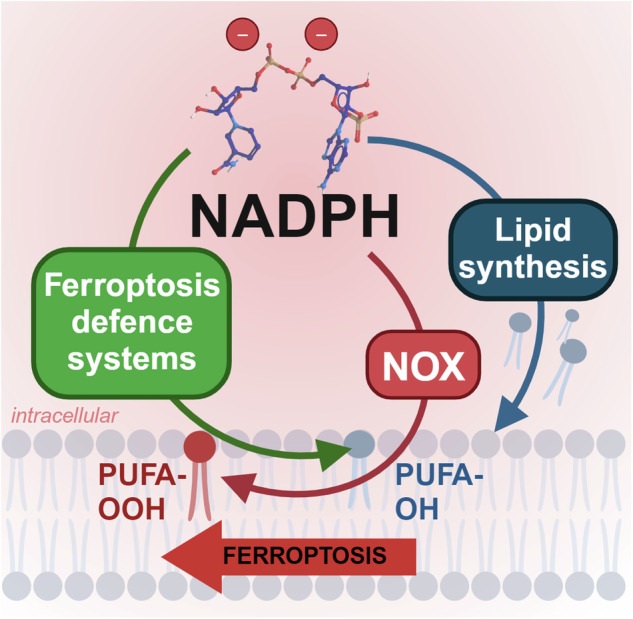
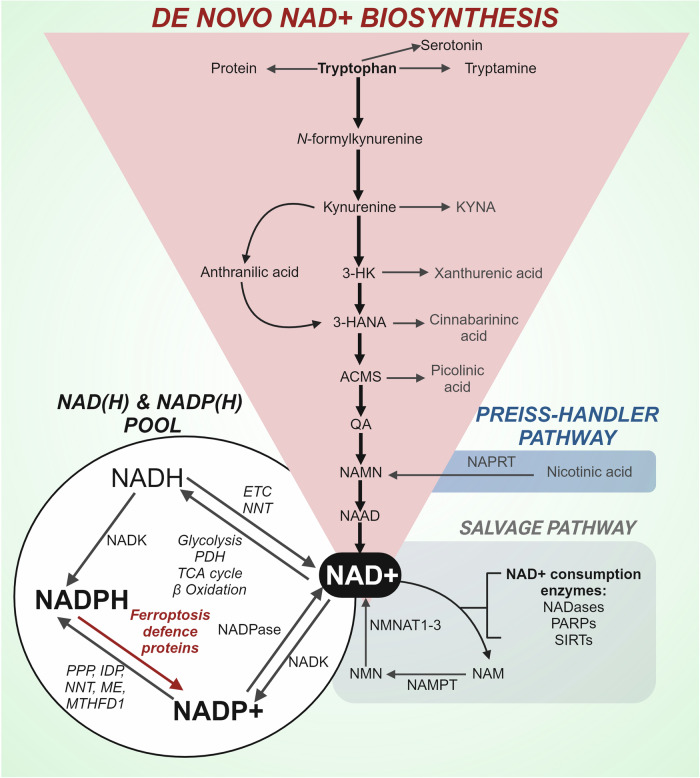
Rampant phospholipid peroxidation initiated by iron causes ferroptosis unless this is restrained by cellular defences. Ferroptosis is increasingly implicated in a host of diseases, and unlike other cell death programs the physiological initiation of ferroptosis is conceived to occur not by an endogenous executioner, but by the withdrawal of cellular guardians that otherwise constantly oppose ferroptosis induction. Here, we profile key ferroptotic defence strategies including iron regulation, phospholipid modulation and enzymes and metabolite systems: glutathione reductase (GR), Ferroptosis suppressor protein 1 (FSP1), NAD(P)H Quinone Dehydrogenase 1 (NQO1), Dihydrofolate reductase (DHFR), retinal reductases and retinal dehydrogenases (RDH) and thioredoxin reductases (TR). A common thread uniting all key enzymes and metabolites that combat lipid peroxidation during ferroptosis is a dependence on a key cellular reductant, nicotinamide adenine dinucleotide phosphate (NADPH). We will outline how cells control central carbon metabolism to produce NADPH and necessary precursors to defend against ferroptosis. Subsequently we will discuss evidence for ferroptosis and NADPH dysregulation in different disease contexts including glucose-6-phosphate dehydrogenase deficiency, cancer and neurodegeneration. Finally, we discuss several anti-ferroptosis therapeutic strategies spanning the use of radical trapping agents, iron modulation and glutathione dependent redox support and highlight the current landscape of clinical trials focusing on ferroptosis.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: Ashley Bush is an Associate Editor of Signal Transduction and Targeted Therapy, but he has not been involved in the process of the manuscript handling. All the other authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
