

Robust proteome profiling of cysteine-reactive fragments using label-free chemoproteomics
- PMID: 39746958
- PMCID: PMC11697256
- DOI: 10.1038/s41467-024-55057-5
Robust proteome profiling of cysteine-reactive fragments using label-free chemoproteomics
Abstract
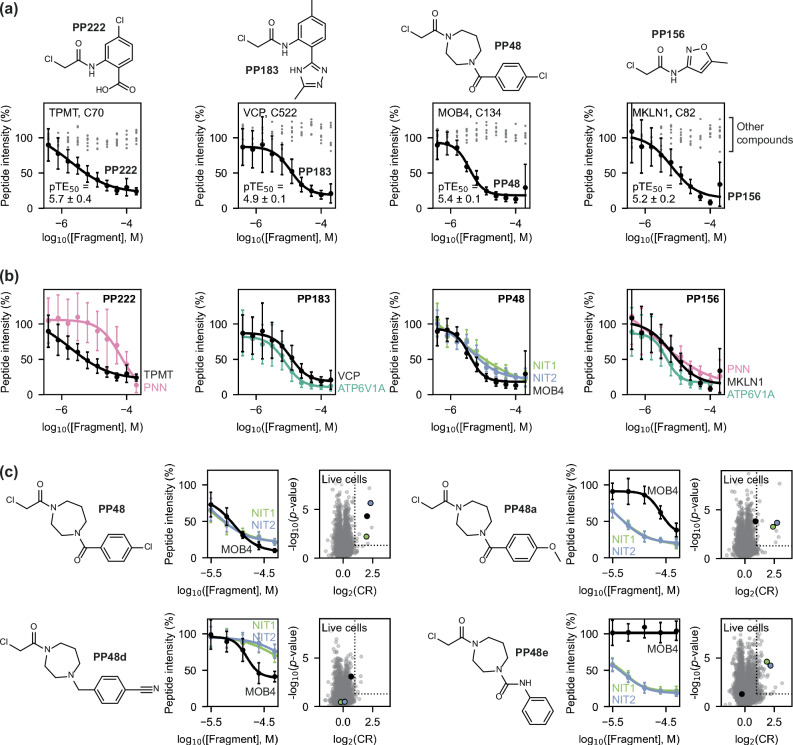
Identifying pharmacological probes for human proteins represents a key opportunity to accelerate the discovery of new therapeutics. High-content screening approaches to expand the ligandable proteome offer the potential to expedite the discovery of novel chemical probes to study protein function. Screening libraries of reactive fragments by chemoproteomics offers a compelling approach to ligand discovery, however, optimising sample throughput, proteomic depth, and data reproducibility remains a key challenge. We report a versatile, label-free quantification proteomics platform for competitive profiling of cysteine-reactive fragments against the native proteome. This high-throughput platform combines SP4 plate-based sample preparation with rapid chromatographic gradients. Data-independent acquisition performed on a Bruker timsTOF Pro 2 consistently identified ~23,000 cysteine sites per run, with a total of ~32,000 cysteine sites profiled in HEK293T and Jurkat lysate. Crucially, this depth in cysteinome coverage is met with high data completeness, enabling robust identification of liganded proteins. In this study, 80 reactive fragments were screened in two cell lines identifying >400 ligand-protein interactions. Hits were validated through concentration-response experiments and the platform was utilised for hit expansion and live cell experiments. This label-free platform represents a significant step forward in high-throughput proteomics to evaluate ligandability of cysteines across the human proteome.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Garbaccio, R. M. & Parmee, E. R. The impact of chemical probes in drug discovery: a pharmaceutical industry perspective. Cell Chem. Biol.23, 10–17 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
