

Constitutive activation of the Src-family kinases Fgr and Hck enhances the tumor burden of acute myeloid leukemia cells in immunocompromised mice
- PMID: 39747387
- PMCID: PMC11697302
- DOI: 10.1038/s41598-024-83740-6
Constitutive activation of the Src-family kinases Fgr and Hck enhances the tumor burden of acute myeloid leukemia cells in immunocompromised mice
Abstract
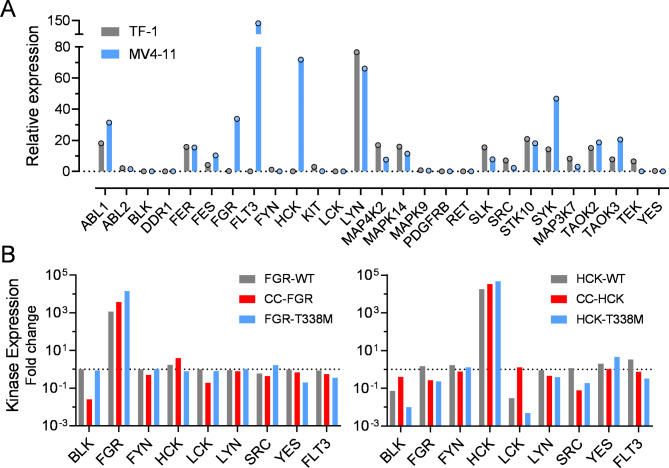
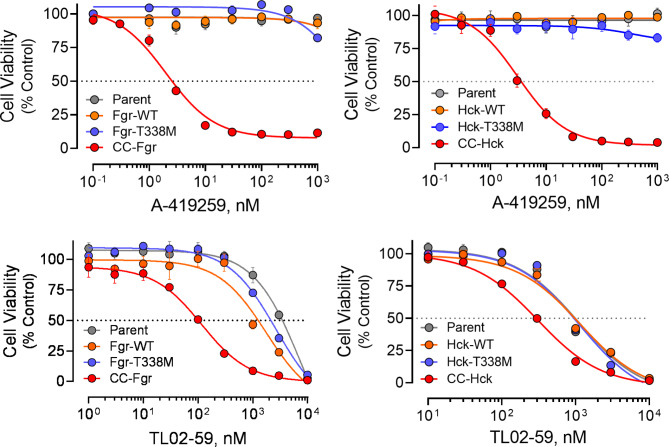
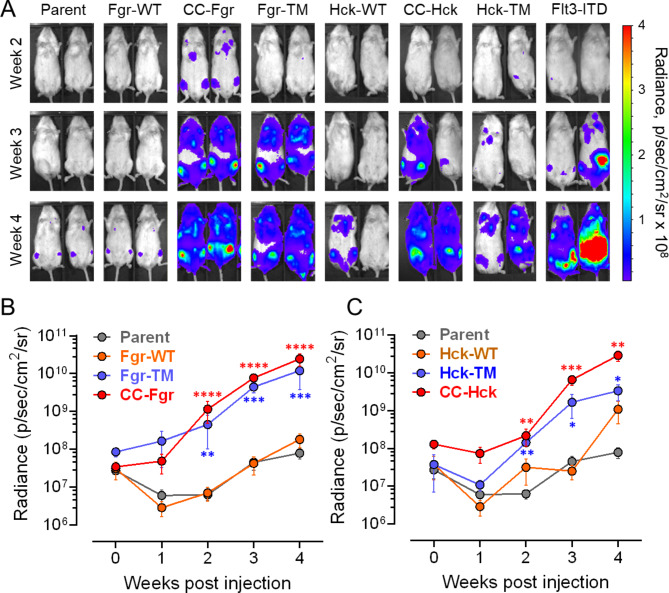
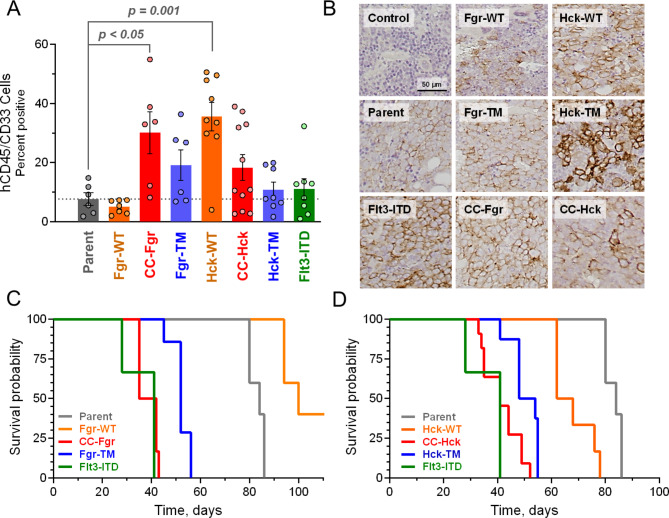
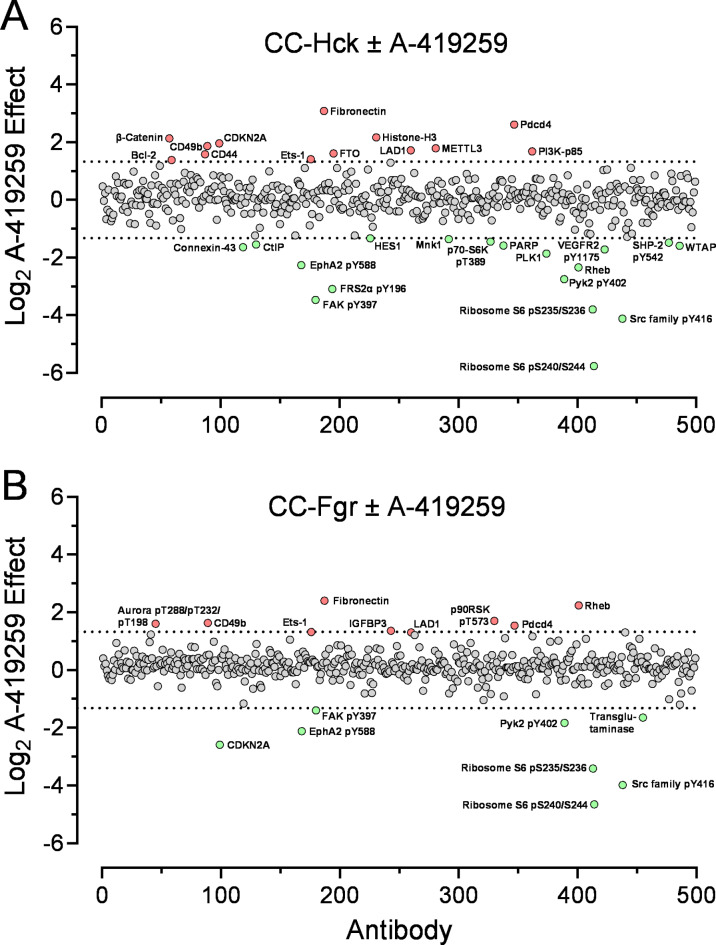
Overexpression of the myeloid Src-family kinases Fgr and Hck has been linked to the development of acute myeloid leukemia (AML). Here we characterized the contribution of active forms of these kinases to AML cell cytokine dependence, inhibitor sensitivity, and AML cell engraftment in vivo. The human TF-1 erythroleukemia cell line was used as a model system as it does not express endogenous Hck or Fgr. To induce constitutive kinase activity, Hck and Fgr were fused to the coiled-coil (CC) oligomerization domain of the breakpoint cluster region protein associated with the Bcr-Abl tyrosine kinase in chronic myeloid leukemia. Expression of CC-Hck or CC-Fgr transformed TF-1 cells to a granulocyte-macrophage colony-stimulating factor (GM-CSF)-independent phenotype that correlated with enhanced phosphorylation of the kinase domain activation loop. Both CC-Hck and CC-Fgr cell populations became sensitized to growth arrest by Src-family kinase inhibitors previously shown to suppress the growth of bone marrow cells from AML patients in vitro and decrease AML cell engraftment in immunocompromised mice. Methionine substitution of the 'gatekeeper' residue (Thr338) also stimulated Hck and Fgr kinase activity and transformed TF-1 cells to GM-CSF independence without CC fusion. TF-1 cells expressing either active form of Hck or Fgr engrafted immunocompromised mice faster and developed more extensive tumors compared to mice engrafted with the parent cell line, resulting in shorter survival. Expression of wild-type Hck also significantly enhanced bone marrow engraftment without an activating mutation. Reverse phase protein array analysis linked active Hck and Fgr to the mammalian target of rapamycin complex-1/p70 S6 ribosomal protein (mTORC-1/S6) kinase and focal adhesion kinase (Fak) signaling pathways. Combining Hck and Fgr inhibitors with existing mTORC-1/S6 kinase or Fak inhibitors may improve clinical responses and reduce the potential for acquired resistance.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare that they have no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
