

The impact of common and rare genetic variants on bradyarrhythmia development
- PMID: 39747593
- PMCID: PMC11735381
- DOI: 10.1038/s41588-024-01978-2
The impact of common and rare genetic variants on bradyarrhythmia development
Abstract
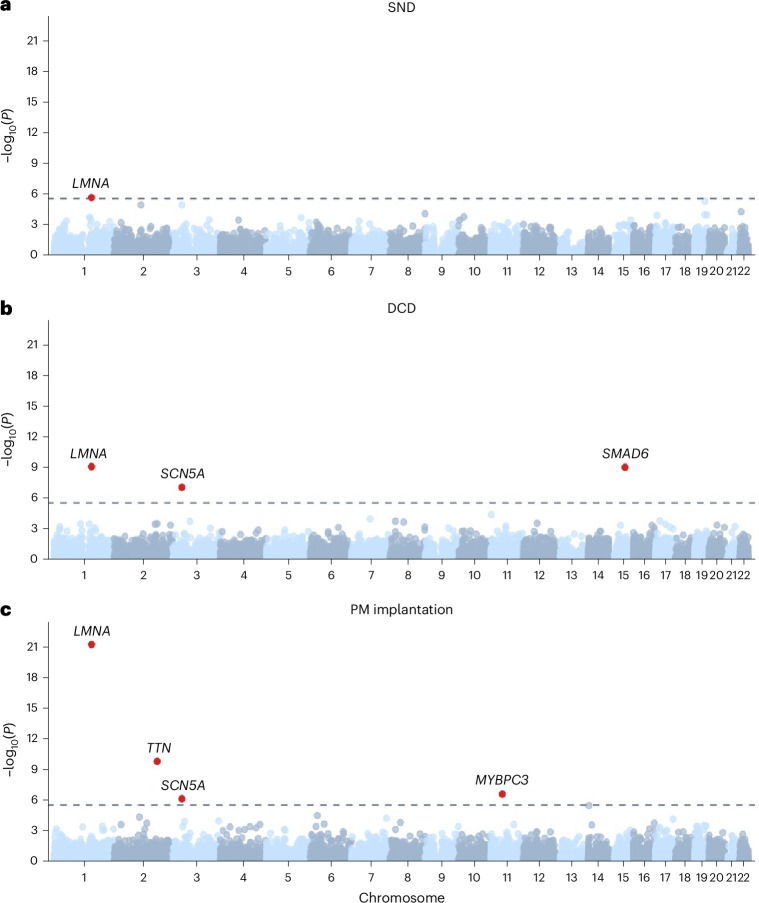
To broaden our understanding of bradyarrhythmias and conduction disease, we performed common variant genome-wide association analyses in up to 1.3 million individuals and rare variant burden testing in 460,000 individuals for sinus node dysfunction (SND), distal conduction disease (DCD) and pacemaker (PM) implantation. We identified 13, 31 and 21 common variant loci for SND, DCD and PM, respectively. Four well-known loci (SCN5A/SCN10A, CCDC141, TBX20 and CAMK2D) were shared for SND and DCD, while others were more specific for SND or DCD. SND and DCD showed a moderate genetic correlation (rg = 0.63). Cardiomyocyte-expressed genes were enriched for contributions to DCD heritability. Rare-variant analyses implicated LMNA for all bradyarrhythmia phenotypes, SMAD6 and SCN5A for DCD and TTN, MYBPC3 and SCN5A for PM. These results show that variation in multiple genetic pathways (for example, ion channel function, cardiac developmental programs, sarcomeric structure and cellular homeostasis) appear critical to the development of bradyarrhythmias.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.A.L. is employed by Novartis as of July 2022. S.A.L. received sponsored research support from Bristol Myers Squibb/Pfizer, Bayer AG, Boehringer Ingelheim, Fitbit, IBM, Medtronic and Premier and consulted for Bristol Myers Squibb/Pfizer, Bayer AG, Blackstone Life Sciences and Invitae. C.R. is a full-time employee at GSK as of July 2024. P.T.E. has received sponsored research support from Bayer AG, IBM Research, Bristol Myers Squibb, Pfizer and Novo Nordisk; he has also served on advisory boards or consulted for Bayer AG and MyoKardia. S.M.D. receives research support from RenalytixAI and Novo Nordisk, outside the scope of the current research. The FinnGen project is funded by two grants from Business Finland (HUS 4685/31/2016 and UH 4386/31/2016) and the following industry partners: AbbVie, AstraZeneca, Biogen, Bristol Myers Squibb (and Celgene and Celgene International II Sàrl), Genentech, Merck Sharp & Dohme, Pfizer, GlaxoSmithKline Intellectual Property Development, Sanofi US Services, Maze Therapeutics, Janssen Biotech, Novartis AG and Boehringer Ingelheim International GmbH. B.M.P. serves on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson. The remaining authors declare no competing interests.
Figures







References
-
- Greenspon, A. J. et al. Trends in permanent pacemaker implantation in the United States from 1993 to 2009: increasing complexity of patients and procedures. J. Am. Coll. Cardiol.60, 1540–1545 (2012). - PubMed
-
- Veldkamp, M. W. et al. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ. Res.92, 976–983 (2003). - PubMed
MeSH terms
Substances
Grants and funding
- R01 HL141901/HL/NHLBI NIH HHS/United States
- R01 HL139738/HL/NHLBI NIH HHS/United States
- 18SFRN34250007/American Heart Association (American Heart Association, Inc.)
- TNE FANTASY 19CV03/Fondation Leducq
- R01 HL092577/HL/NHLBI NIH HHS/United States
- 18SFRN34110082/American Heart Association (American Heart Association, Inc.)
- R01 HL105756/HL/NHLBI NIH HHS/United States
- R01 HL157635/HL/NHLBI NIH HHS/United States
- R01 HL163987/HL/NHLBI NIH HHS/United States
- R01 HL139731/HL/NHLBI NIH HHS/United States
- U01 AG068221/AG/NIA NIH HHS/United States
- 23CDA1050571/American Heart Association (American Heart Association, Inc.)
- T32 HL007101/HL/NHLBI NIH HHS/United States
- 18SFRN34230127/American Heart Association (American Heart Association, Inc.)
- IK2 CX001780/CX/CSRD VA/United States
- 75N92019D00031/HL/NHLBI NIH HHS/United States
- K23 HL169839/HL/NHLBI NIH HHS/United States
- 03-007-2022-0035/Hartstichting (Dutch Heart Foundation)
LinkOut - more resources
Full Text Sources
Miscellaneous
