

The spatially informed mFISHseq assay resolves biomarker discordance and predicts treatment response in breast cancer
- PMID: 39747865
- PMCID: PMC11696812
- DOI: 10.1038/s41467-024-55583-2
The spatially informed mFISHseq assay resolves biomarker discordance and predicts treatment response in breast cancer
Abstract
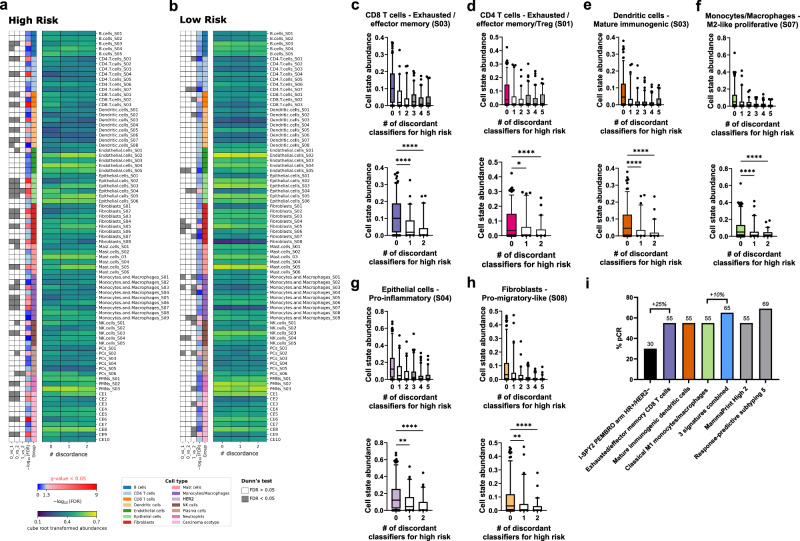
Current assays fail to address breast cancer's complex biology and accurately predict treatment response. On a retrospective cohort of 1082 female breast tissues, we develop and validate mFISHseq, which integrates multiplexed RNA fluorescent in situ hybridization with RNA-sequencing, guided by laser capture microdissection. This technique ensures tumor purity, unbiased whole transcriptome profiling, and explicitly quantifies intratumoral heterogeneity. Here we show mFISHseq has 93% accuracy compared to immunohistochemistry. Our consensus subtyping and risk groups mitigate single sample discordance, provide early and late prognostic information, and identify high risk patients with enriched immune signatures, which predict response to neoadjuvant immunotherapy in the multicenter, phase II, prospective I-SPY2 trial. We identify putative antibody-drug conjugate (ADC)-responsive patients, as evidenced by a 19-feature T-DM1 classifier, validated on I-SPY2. Deploying mFISHseq as a research-use only test on 48 patients demonstrates clinical feasibility, revealing insights into the efficacy of targeted therapies, like CDK4/6 inhibitors, immunotherapies, and ADCs.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: E.D.P., B.H., N.Val., N.M., D.G., S.G., H.I., T.O., M.G., L.B., S.B., J.B., K.B., D.D., Z.K., M.K., D.L., V.Mam., V.Man., N.Voj., M.R. and P.Č. are current, or former employees of MultiplexDX, a biotechnology company that is developing a lab developed diagnostic test called Multiplex8+ ( https://www.multiplexdx.com/products/multiplex-eight-plus ), which is based on the research presented in the manuscript. P.Č. and E.D.P. are inventors and MultiplexDX, s.r.o is the assignee on patent applications that were filed in relation to the technology and research outlined in the manuscript. These include a family of patents entitled “METHOD FOR DIAGNOSING DISEASES USING MULTIPLEX FLUORESCENCE AND SEQUENCING” (WO/2020/070325, EP3775277, CA3114689, AU2019354863, SG11202103466T, KR1020210071003, CN113366118, BR112021006454, US20230037279, JP2022513333, IL282067, and NZ774986) as well as submitted EPO and PCT patents that are not published. T.T., F.P., and J.N.K. are members of the Scientific Advisory board at MultiplexDX. F.P. reports consulting services and serving on the advisory board for AstraZeneca. J.N.K. declares consulting services for Owkin, France, DoMore Diagnostics, Norway, Panakeia, UK, Scailyte, Switzerland, Cancilico, Germany, Mindpeak, Germany, and Histofy, UK; furthermore, he holds shares in StratifAI GmbH, Germany, has received a research grant by GSK, and has received honoraria by AstraZeneca, Bayer, Eisai, Janssen, MSD, BMS, Roche, Pfizer and Fresenius. All other authors declare no competing interests. Ethics & Inclusions: This study complies with all relevant ethical regulations for human research participants in accordance with the Declaration of Helsinki. The collection and use of human tissue in the retrospective clinical validation study on 1082 patients and the RUO testing cohort of 48 patients were approved by the Ethics Committee of the Bratislava Self-Governing Region (Ref. No. 05320/2020/HF). Additionally, the retrospective cohort received approval from the Ethics Commission of the Medical University of Graz on behalf of Biobank Graz (No. 34-354 ex 21/21, 1158−2022). Informed consent was obtained for tissues in the retrospective study from tissue sources including biobanks (PATH Biobank and Biobank Graz), a hospital (Malaga), and commercial companies (AMSBio and Precision for Medicine). For the RUO cohort, all 48 patients signed informed consent forms, with their oncologists approving them prior to tissue processing. The RUO testing occurred through routine testing with collaborating hospitals in Slovakia and included consultations and participation of local oncologists and researchers for patient selection, collecting clinicopathological data, obtaining informed consent, and explaining results. No discrimination occurred in the selection of patients for RUO testing and both clinical partners and MultiplexDX ensured sensitive patient information was anonymized when appropriate and secured both physically and electronically. A local pathologist (Dr. Karol Kajo) was also consulted with in both the retrospective and RUO cohorts to identify invasive breast cancer or unique histology in more challenging cases.
Figures








Update of
-
Multiplexed RNA-FISH-guided Laser Capture Microdissection RNA Sequencing Improves Breast Cancer Molecular Subtyping, Prognostic Classification, and Predicts Response to Antibody Drug Conjugates.medRxiv [Preprint]. 2023 Dec 6:2023.12.05.23299341. doi: 10.1101/2023.12.05.23299341. medRxiv. 2023. Update in: Nat Commun. 2025 Jan 2;16(1):226. doi: 10.1038/s41467-024-55583-2. PMID: 38105959 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
