

Pan-cancer drivers of metastasis
- PMID: 39748426
- PMCID: PMC11697158
- DOI: 10.1186/s12943-024-02182-w
Pan-cancer drivers of metastasis
Abstract
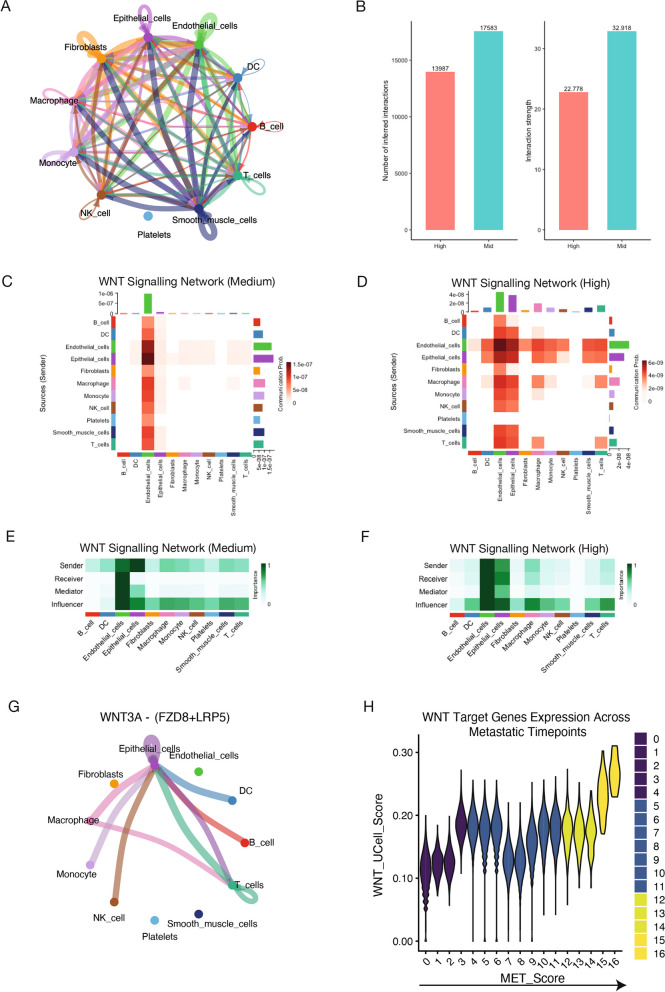
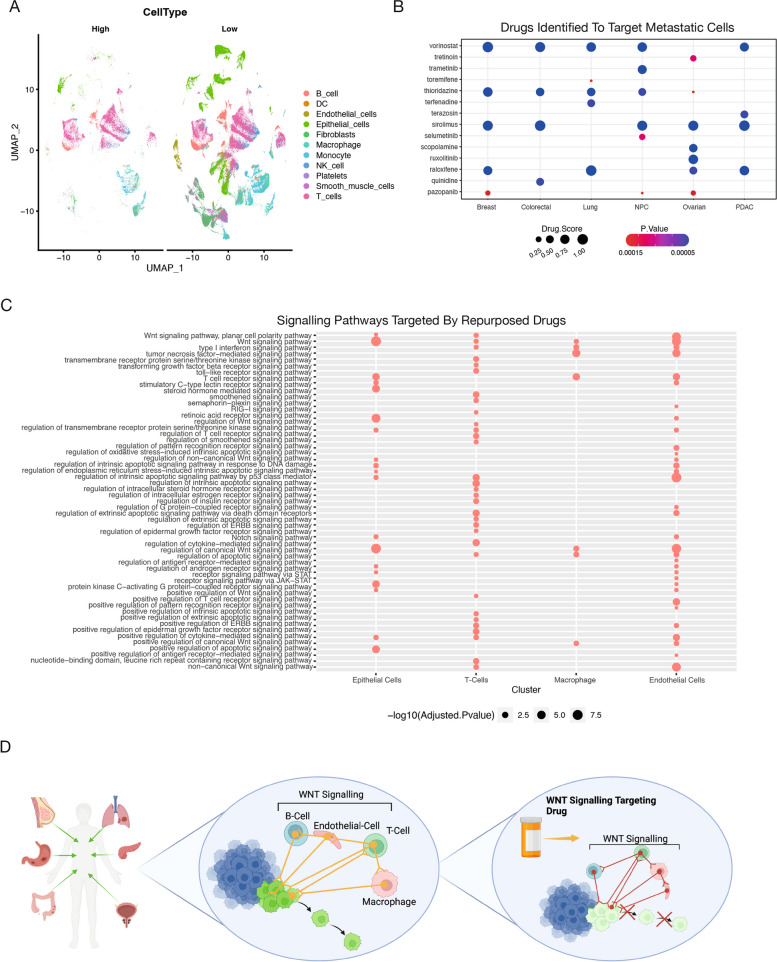
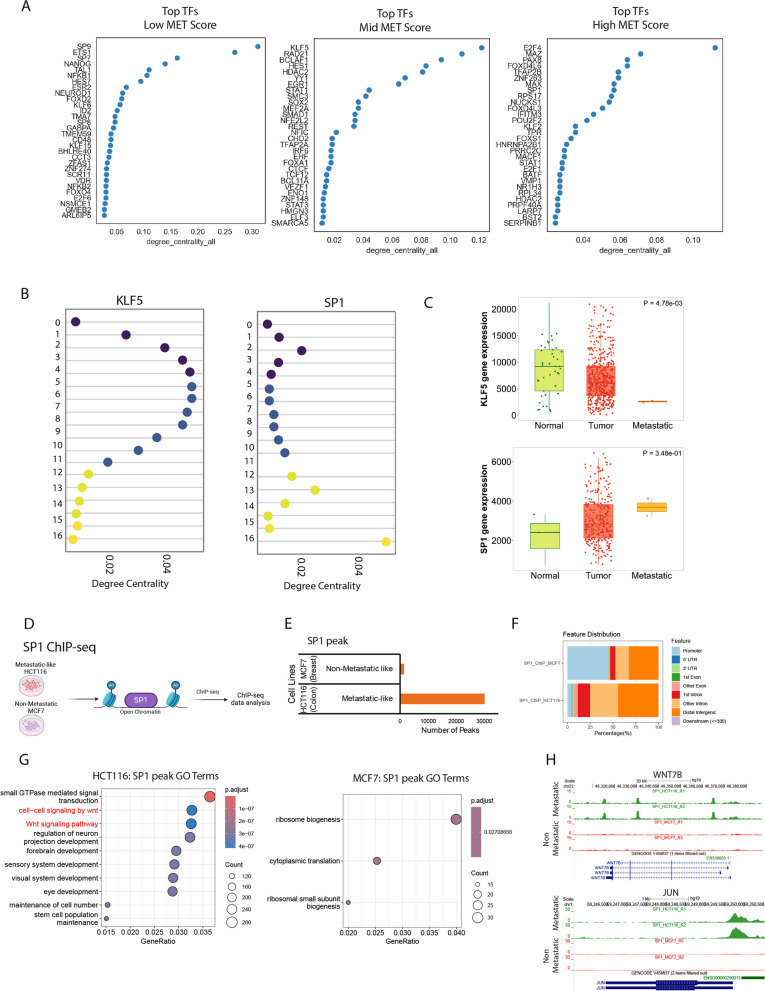
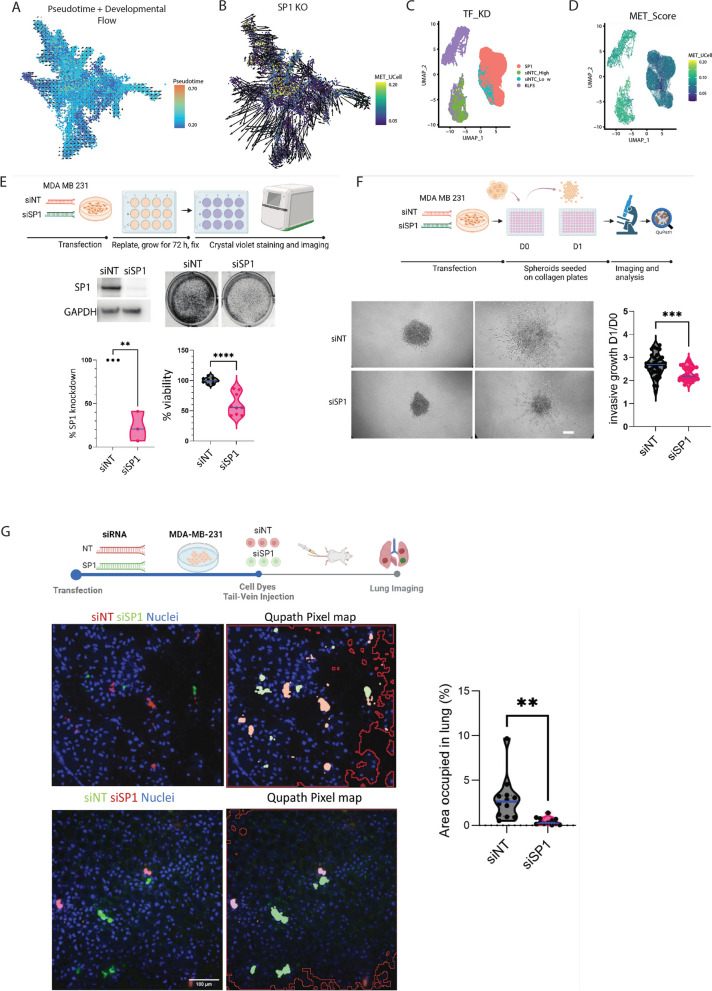
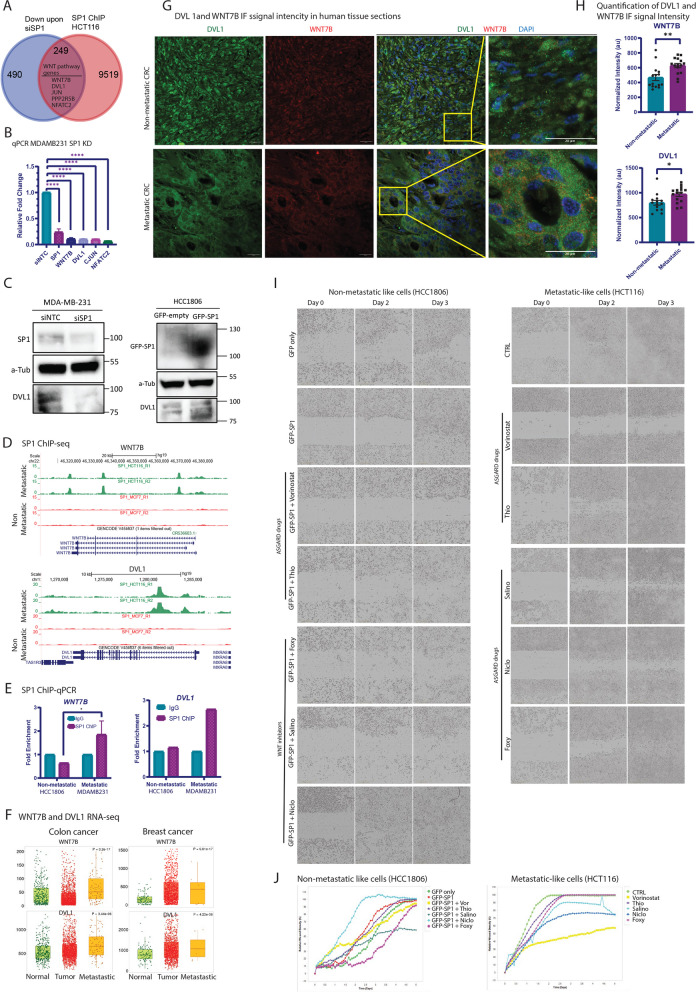
Metastasis remains a leading cause of cancer-related mortality, irrespective of the primary tumour origin. However, the core gene regulatory program governing distinct stages of metastasis across cancers remains poorly understood. We investigate this through single-cell transcriptome analysis encompassing over two hundred patients with metastatic and non-metastatic tumours across six cancer types. Our analysis revealed a prognostic core gene signature that provides insights into the intricate cellular dynamics and gene regulatory networks driving metastasis progression at the pan-cancer and single-cell level. Notably, the dissection of transcription factor networks active across different stages of metastasis, combined with functional perturbation, identified SP1 and KLF5 as key regulators, acting as drivers and suppressors of metastasis, respectively, at critical steps of this transition across multiple cancer types. Through in vivo and in vitro loss of function of SP1 in cancer cells, we revealed its role in driving cancer cell survival, invasive growth, and metastatic colonisation. Furthermore, tumour cells and the microenvironment increasingly engage in communication through WNT signalling as metastasis progresses, driven by SP1. Further validating these observations, a drug repurposing analysis identified distinct FDA-approved drugs with anti-metastasis properties, including inhibitors of WNT signalling across various cancers.
Keywords: Cancer; Gene regulation; Metastasis; Single-cell heterogeneity; Transcription Factors.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Competing interests: The authors declare no competing interests.
Figures
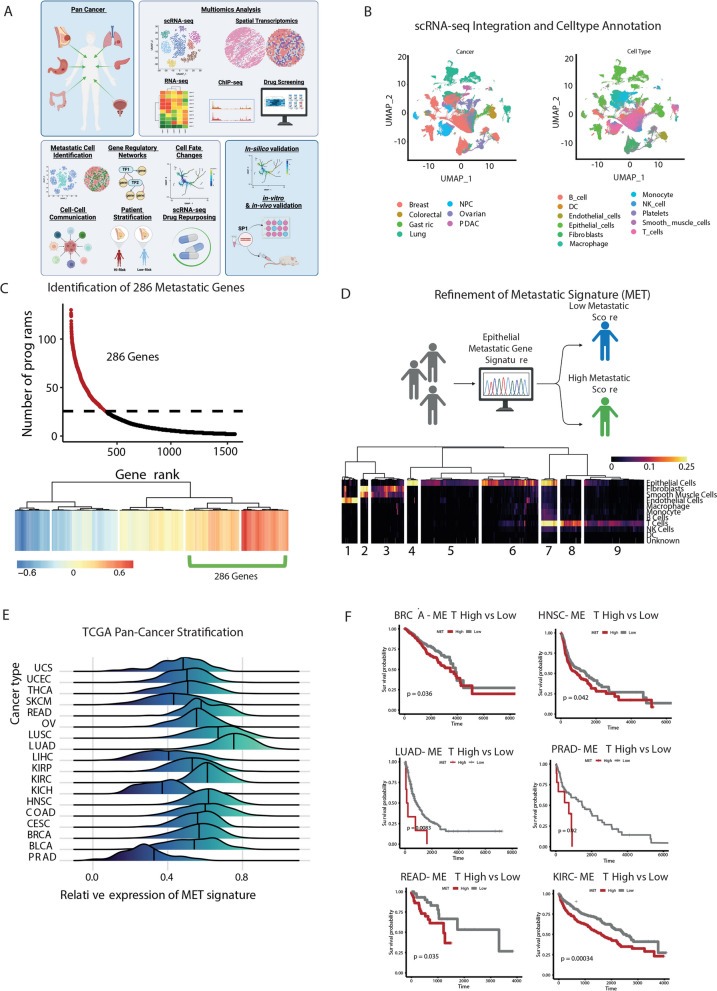
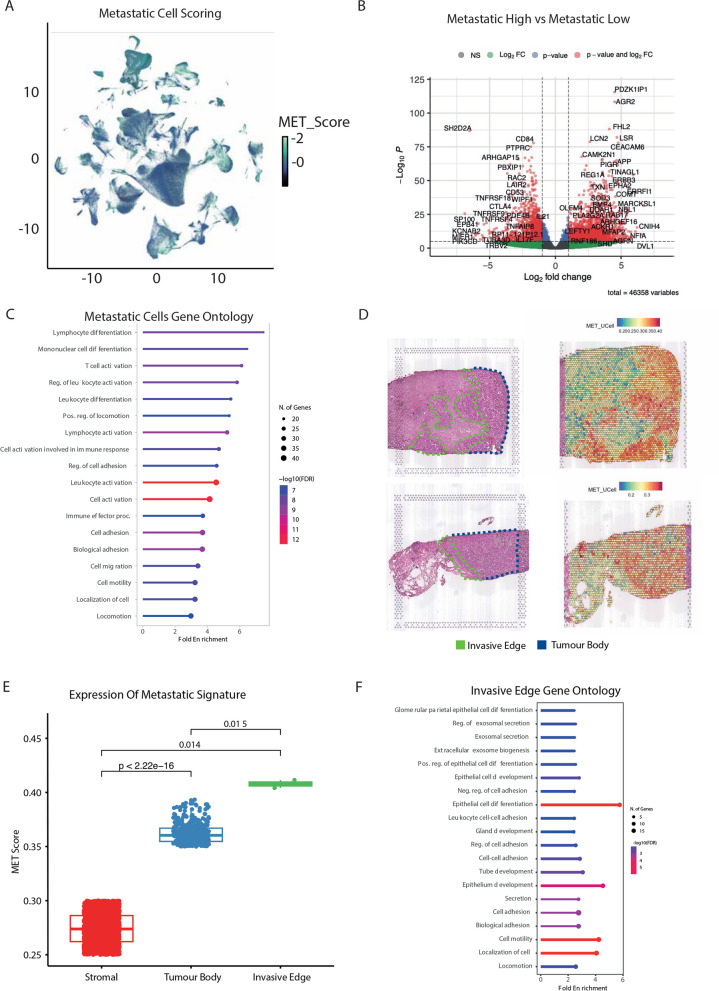
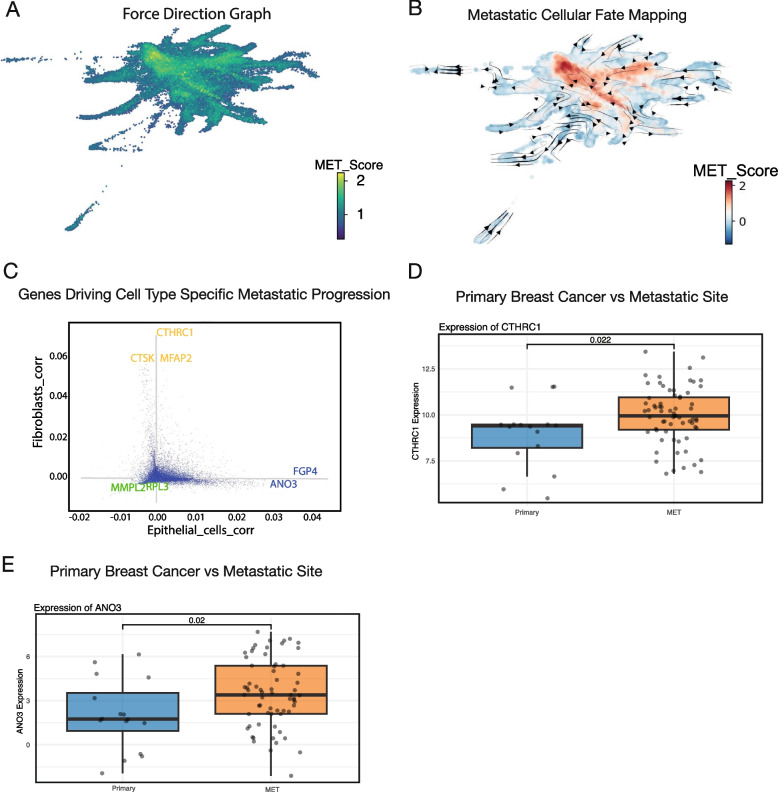
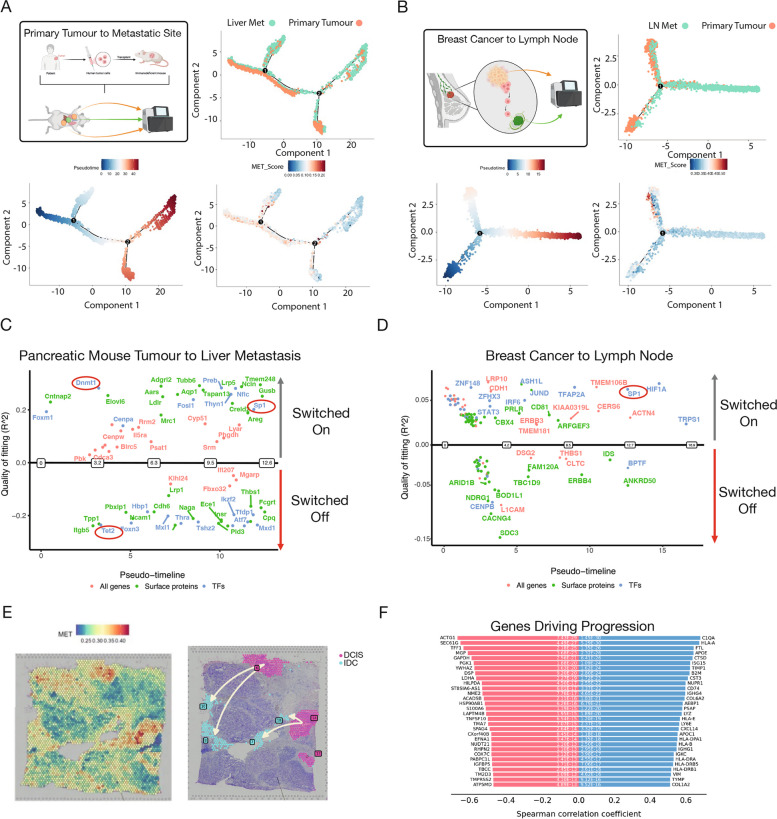
References
-
- Broggio J, Bannister N. Cancer survival by stage at diagnosis for England. Newport, UK Off Natl Stat 2016.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
