

EEFSEC deficiency: A selenopathy with early-onset neurodegeneration
- PMID: 39753114
- PMCID: PMC11739927
- DOI: 10.1016/j.ajhg.2024.12.001
EEFSEC deficiency: A selenopathy with early-onset neurodegeneration
Abstract
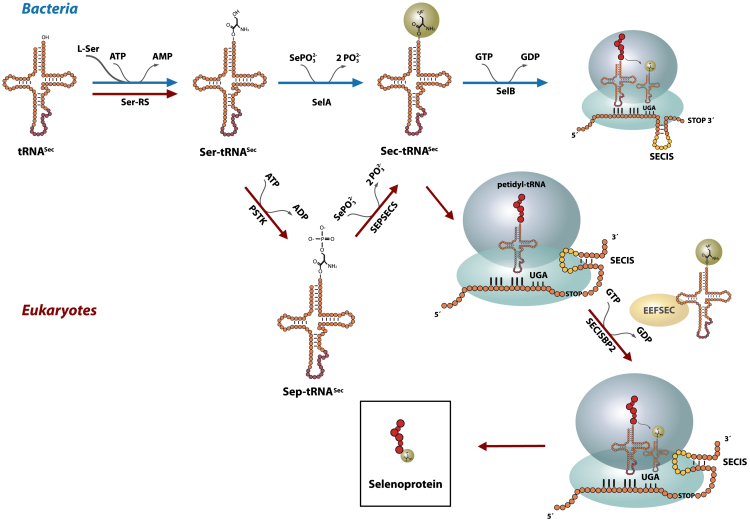
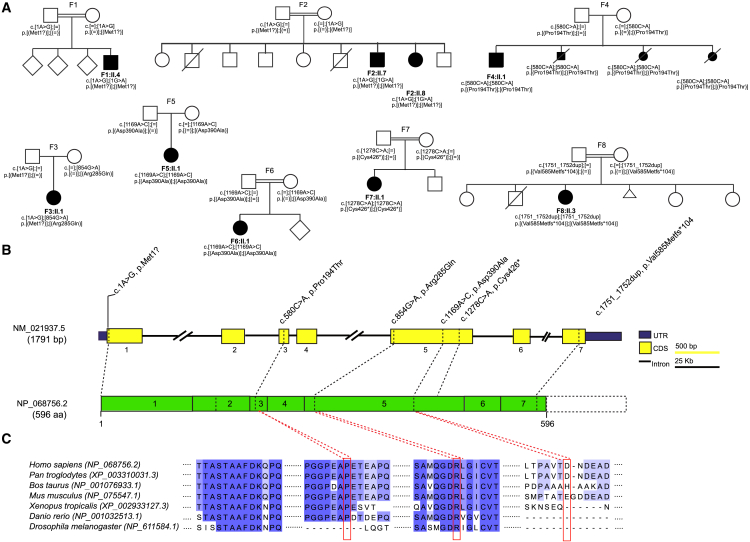
Inborn errors of selenoprotein expression arise from deleterious variants in genes encoding selenoproteins or selenoprotein biosynthetic factors, some of which are associated with neurodegenerative disorders. This study shows that bi-allelic selenocysteine tRNA-specific eukaryotic elongation factor (EEFSEC) variants cause selenoprotein deficiency, leading to progressive neurodegeneration. EEFSEC deficiency, an autosomal recessive disorder, manifests with global developmental delay, progressive spasticity, ataxia, and seizures. Cerebral MRI primarily demonstrated a cerebellar pathology, including hypoplasia and progressive atrophy. Exome or genome sequencing identified six different bi-allelic EEFSEC variants in nine individuals from eight unrelated families. These variants showed reduced EEFSEC function in vitro, leading to lower levels of selenoproteins in fibroblasts. In line with the clinical phenotype, an eEFSec-RNAi Drosophila model displays progressive impairment of motor function, which is reflected in the synaptic defects in this model organisms. This study identifies EEFSEC deficiency as an inborn error of selenocysteine metabolism. It reveals the pathophysiological mechanisms of neurodegeneration linked to selenoprotein metabolism, suggesting potential targeted therapies.
Keywords: EEFSEC deficiency; cerebellar atrophy; cerebellar hypoplasia; epilepsy; progressive spasticity; selenopathy; selenoproteins.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.B. is a co-founder, shareholder, and CTO of AIRAmed GmbH.
Figures



References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
