

Context-dependent effects of CDKN2A and other 9p21 gene losses during the evolution of esophageal cancer
- PMID: 39753721
- PMCID: PMC11779637
- DOI: 10.1038/s43018-024-00876-0
Context-dependent effects of CDKN2A and other 9p21 gene losses during the evolution of esophageal cancer
Abstract
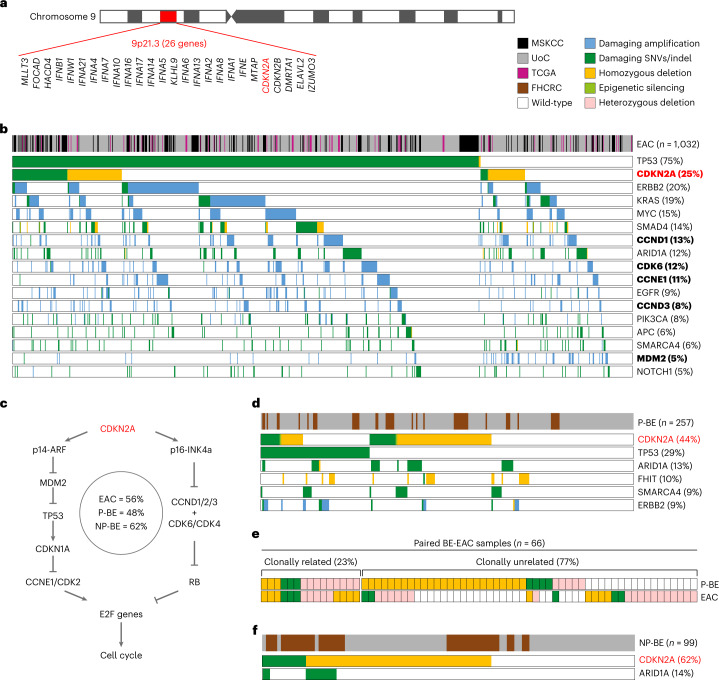
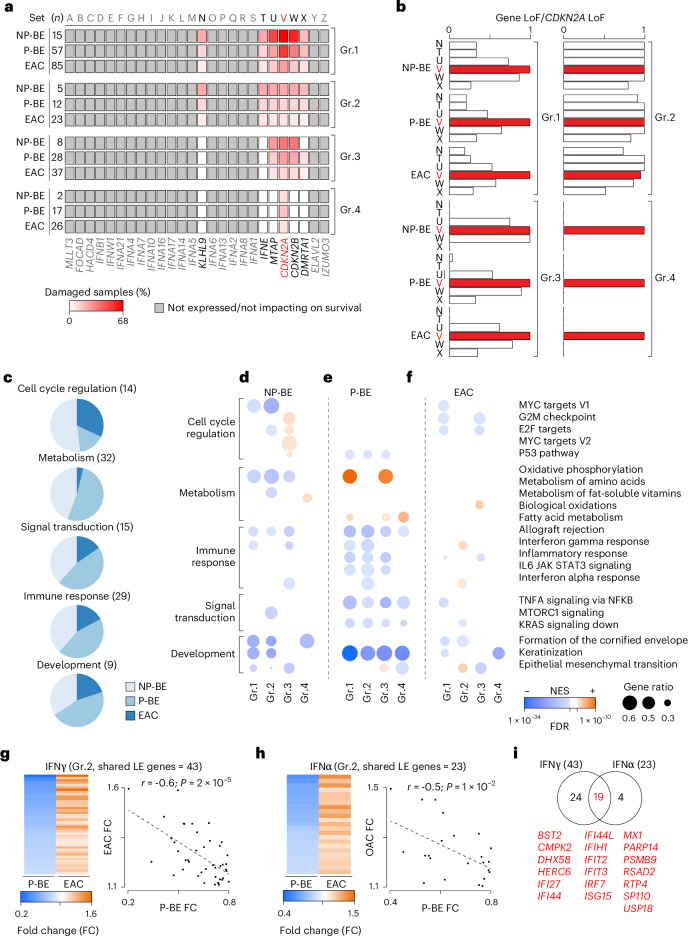
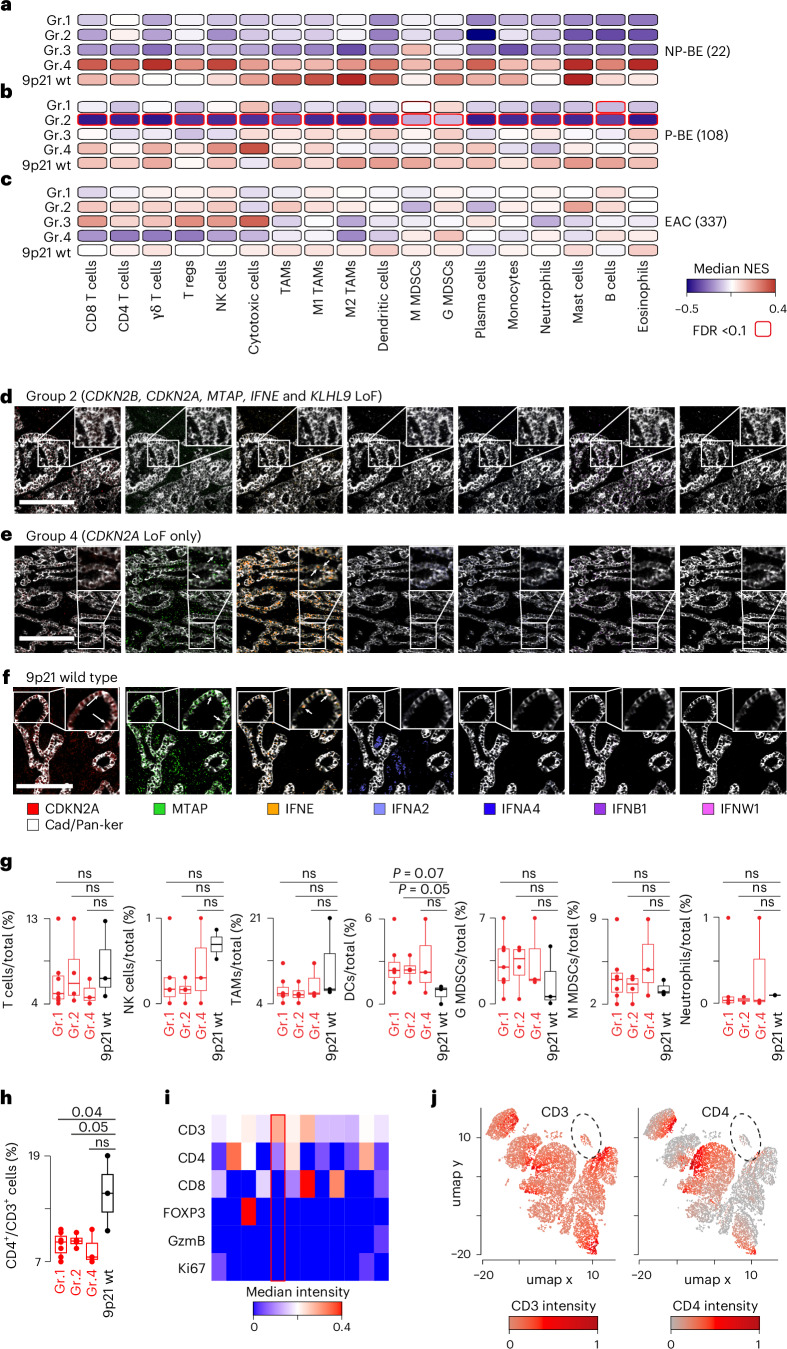
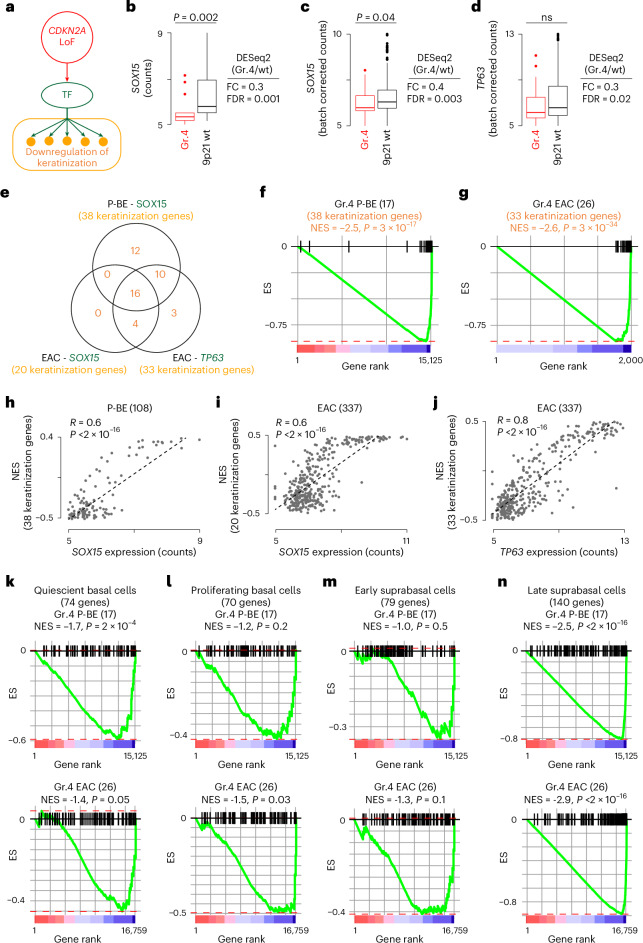
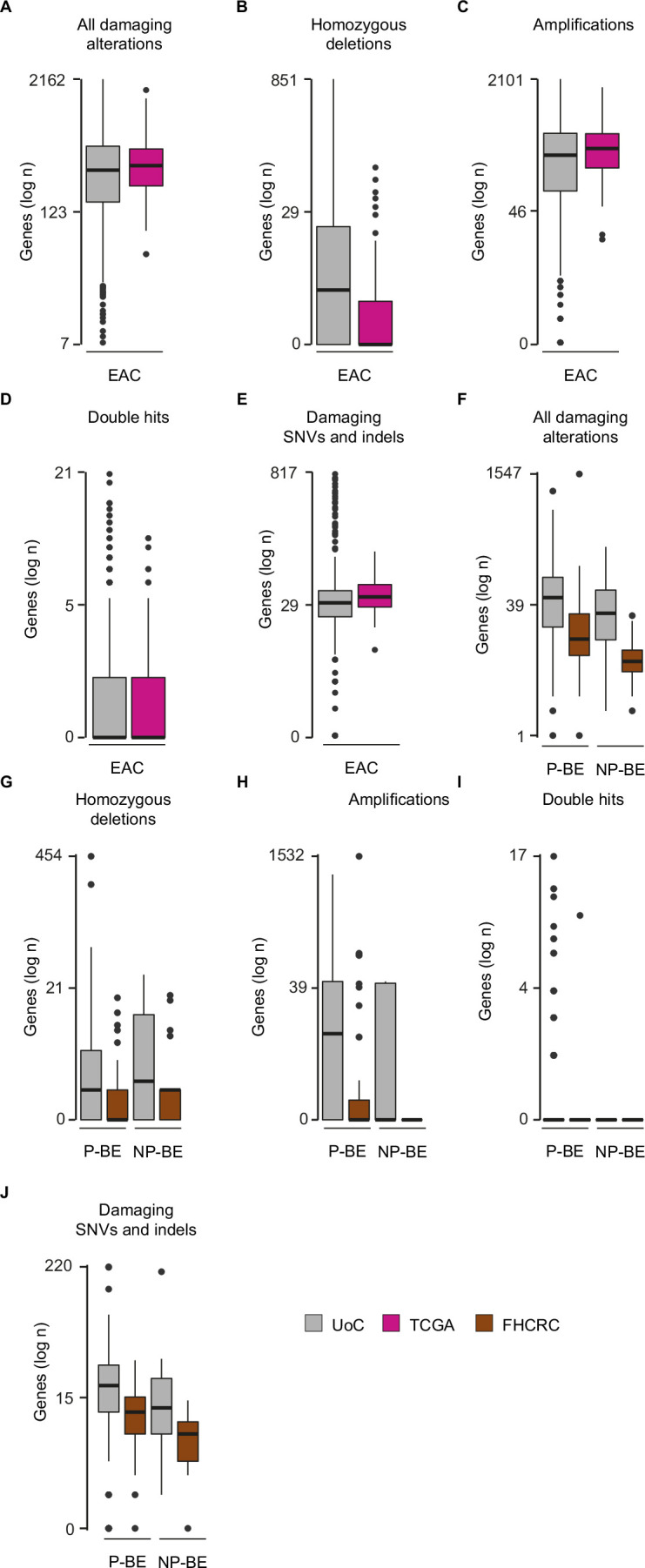
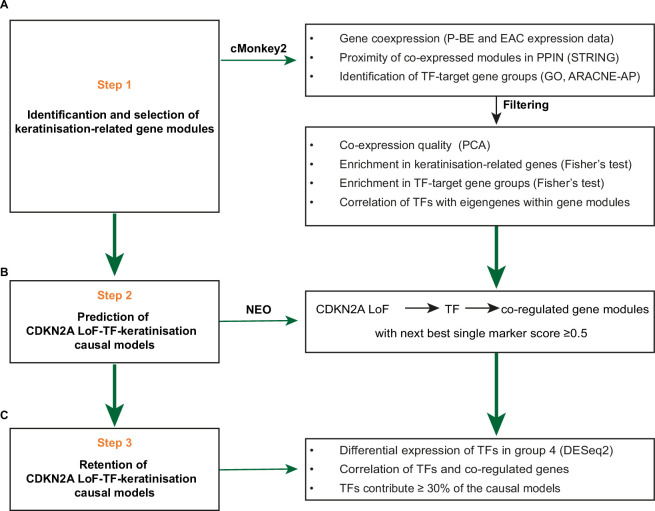
CDKN2A is a tumor suppressor located in chromosome 9p21 and frequently lost in Barrett's esophagus (BE) and esophageal adenocarcinoma (EAC). How CDKN2A and other 9p21 gene co-deletions affect EAC evolution remains understudied. We explored the effects of 9p21 loss in EACs and cancer progressor and non-progressor BEs with matched genomic, transcriptomic and clinical data. Despite its cancer driver role, CDKN2A loss in BE prevents EAC initiation by counterselecting subsequent TP53 alterations. 9p21 gene co-deletions predict poor patient survival in EAC but not BE through context-dependent effects on cell cycle, oxidative phosphorylation and interferon response. Immune quantifications using bulk transcriptome, RNAscope and high-dimensional tissue imaging showed that IFNE loss reduces immune infiltration in BE, but not EAC. Mechanistically, CDKN2A loss suppresses the maintenance of squamous epithelium, contributing to a more aggressive phenotype. Our study demonstrates context-dependent roles of cancer genes during disease evolution, with consequences for cancer detection and patient management.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.C.F. is named on patents related to Cytosponge and related assays which have been licensed by the Medical Research Council to Covidien GI Solutions (now Medtronic) and is a co-founder and shareholder (<3%) of CYTED Ltd. The Fitzgerald lab also has an ongoing collaboration with AstraZeneca. The other authors declare no competing interests.
Figures




References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
