

Clinicogenomic landscape of pancreatic adenocarcinoma identifies KRAS mutant dosage as prognostic of overall survival
- PMID: 39753968
- PMCID: PMC11835752
- DOI: 10.1038/s41591-024-03362-3
Clinicogenomic landscape of pancreatic adenocarcinoma identifies KRAS mutant dosage as prognostic of overall survival
Abstract
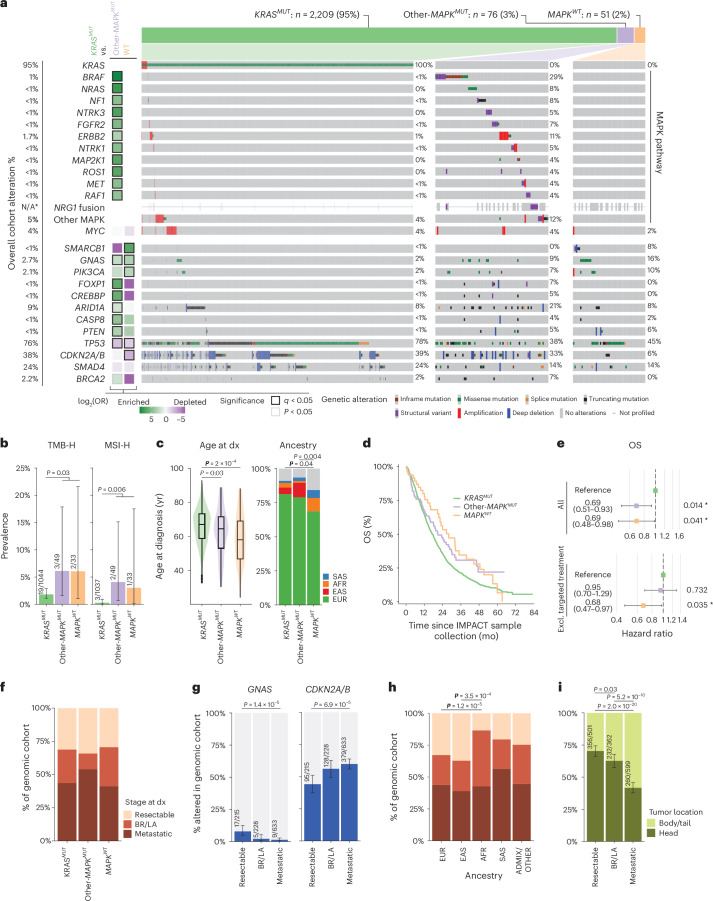
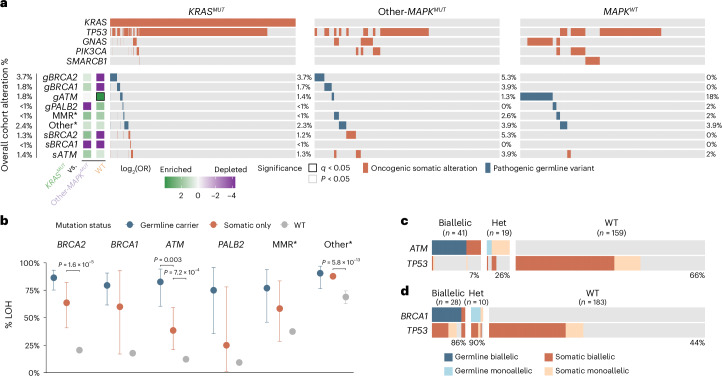
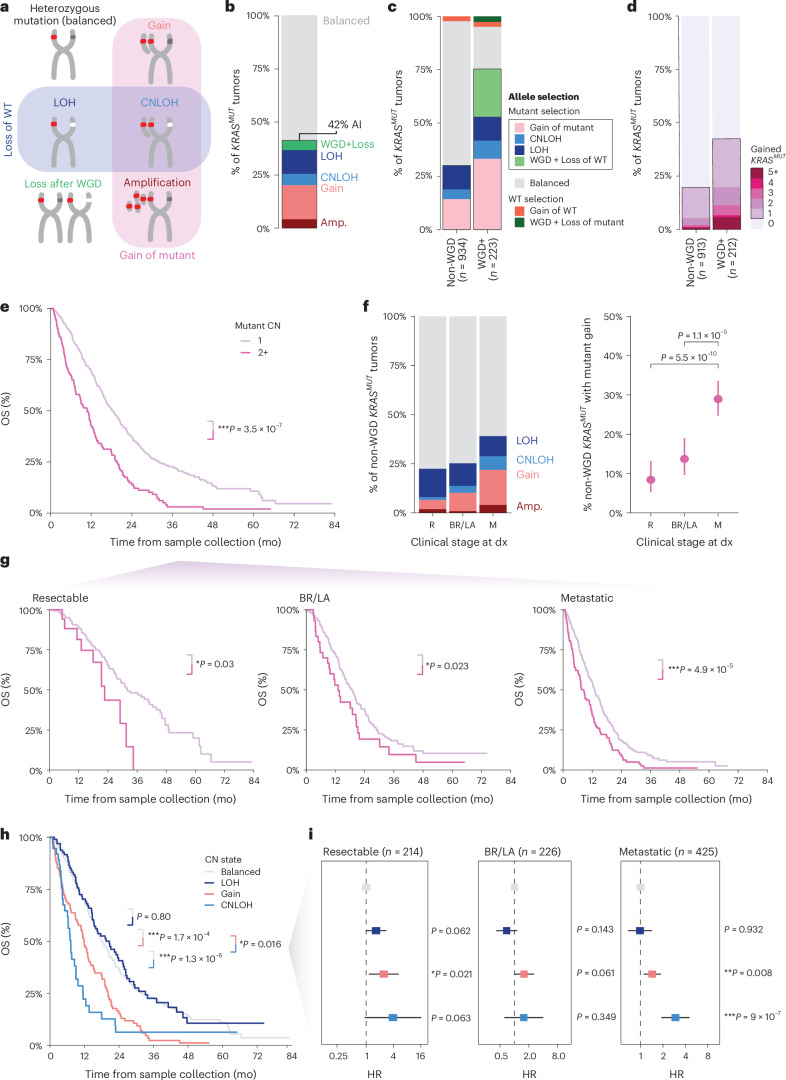
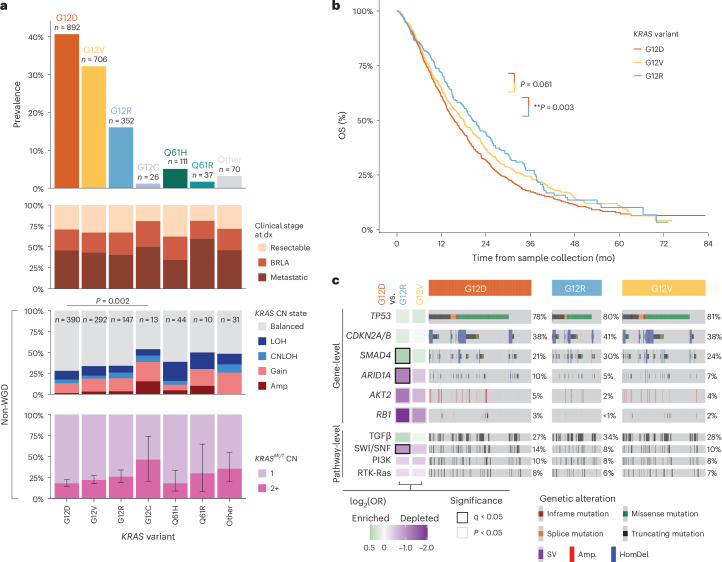
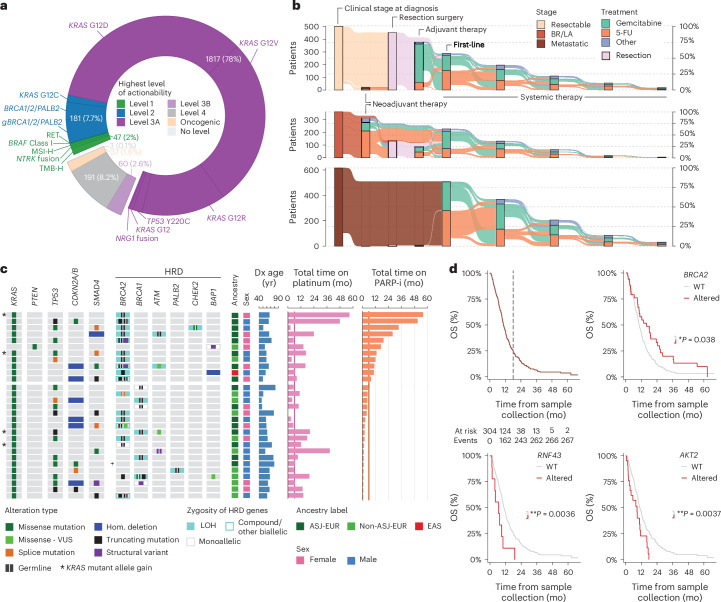
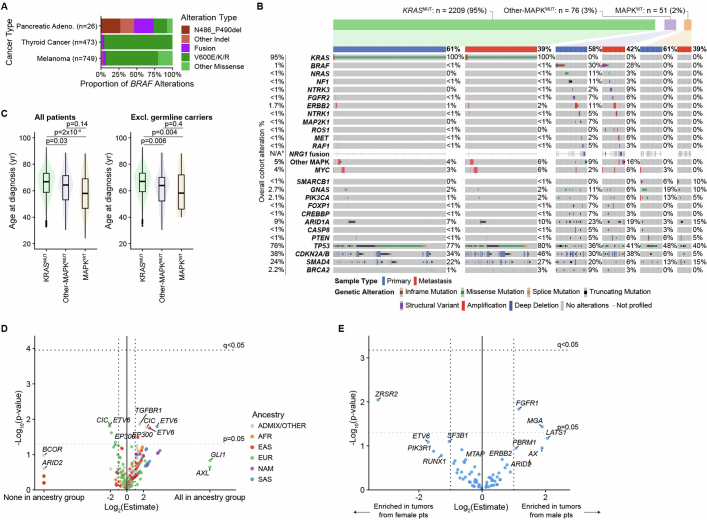
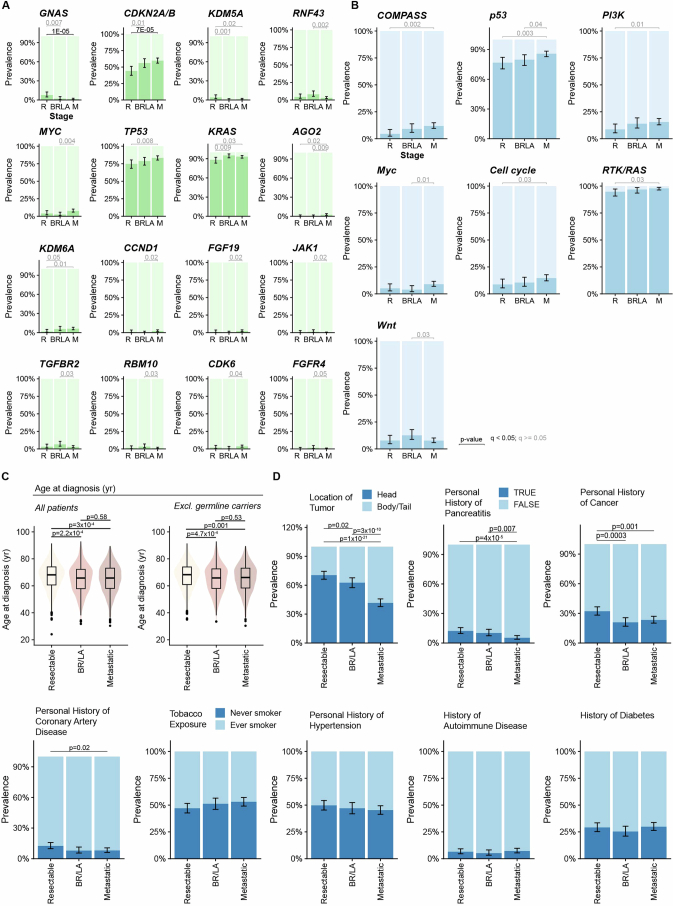
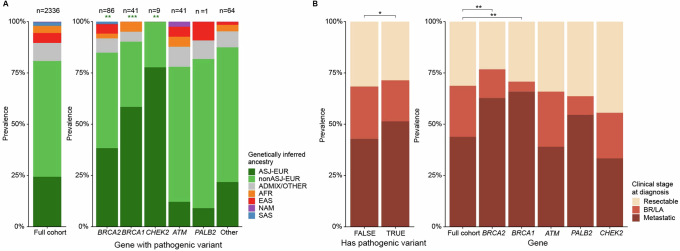
Nearly all pancreatic adenocarcinomas (PDAC) are genomically characterized by KRAS exon 2 mutations. Most patients with PDAC present with advanced disease and are treated with cytotoxic therapy. Genomic biomarkers prognostic of disease outcomes have been challenging to identify. Herein leveraging a cohort of 2,336 patients spanning all disease stages, we characterize the genomic and clinical correlates of outcomes in PDAC. We show that a genomic subtype of KRAS wild-type tumors is associated with early disease onset, distinct somatic and germline features, and significantly better overall survival. Allelic imbalances at the KRAS locus are widespread. KRAS mutant allele dosage gains, observed in one in five (20%) KRAS-mutated diploid tumors, are correlated with advanced disease and demonstrate prognostic potential across disease stages. With the rapidly expanding landscape of KRAS targeting, our findings have potential implications for clinical practice and for understanding de novo and acquired resistance to RAS therapeutics.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.A.P. declares stock ownership in Amgen. A.M.V. declares consulting activity from AstraZeneca (spouse), Eli Lilly (spouse) and Paige AI (spouse), and intellectual property rights (SOPHiA Genetics) (spouse). B.N. is a current employee of Eli Lilly and Company. D. Mandelker declares consulting fees from AstraZeneca. F.B. receives research support from BMS. D.P.K. receives funding from the Thompson Family Foundation and Applebaum Foundation and is a consultant at Merck, BMS, BeiGene, Lilly, Abbvie, Incyte, Janssen, Listen and TG Therapeutics. A.R.B. declares stock ownership in Johnson & Johnson and intellectual property rights in SOPHiA Genetics. W.P. receives research funding from Merck, Astellas, Miracogen, Amgen and Revolution Medicines, is a consultancy/advisory board member for Astellas, EXACT Therapeutics, Innovent Biologics and Regeneron and has received honoraria for CME: American Physician Institute, Integrity. M.F.B. declares consulting activity from AstraZeneca, Eli Lilly and Paige AI and intellectual property rights (SOPHiA Genetics). E.M.O. receives research funding from Agenus, Amgen, Genentech/Roche, BioNTech, AstraZeneca, Arcus, Elicio, Parker Institute, NIH/NCI and Digestive Care, consulting/DSMB role at Arcus, Ability Pharma, Alligator, Agenus, BioNTech, Ipsen, Merck, Moma Therapeutics, Novartis, Syros, Leap Therapeutics, Astellas, BMS, Fibrogen, Revolution Medicine, Merus, Moma Therapeutics and Tango; Agios (spouse), Genentech-Roche (spouse), Eisai (spouse) Servier (spouse). The remaining authors declare no competing interests.
Figures
References
-
- Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin.74, 12–49 (2024). - PubMed
-
- Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med.364, 1817–1825 (2011). - PubMed
-
- Wainberg, Z. A. et al. NAPOLI-3: a randomized, open-label phase 3 study of liposomal irinotecan + 5-fluorouracil/leucovorin + oxaliplatin (NALIRIFOX) versus nab-paclitaxel + gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (mPDAC). J. Clin. Oncol.41, LBA661–LBA661 (2023). - PMC - PubMed
MeSH terms
Substances
Grants and funding
- P50 CA257881/CA/NCI NIH HHS/United States
- R01 CA227534/CA/NCI NIH HHS/United States
- CA008748/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P50 CA257881-01A1/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P30 CA008748/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
