

Protocol for capturing a full transcriptome from single preimplantation embryos using So-Smart-seq
- PMID: 39756032
- PMCID: PMC11758199
- DOI: 10.1016/j.xpro.2024.103540
Protocol for capturing a full transcriptome from single preimplantation embryos using So-Smart-seq
Abstract
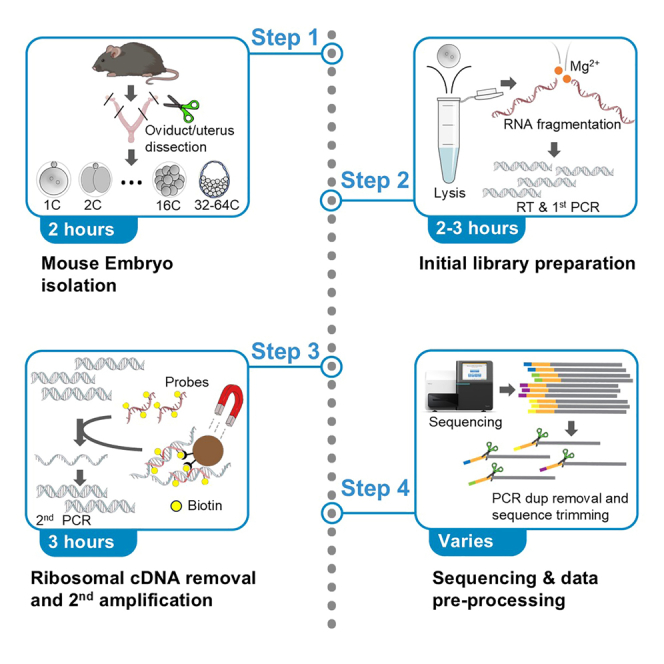
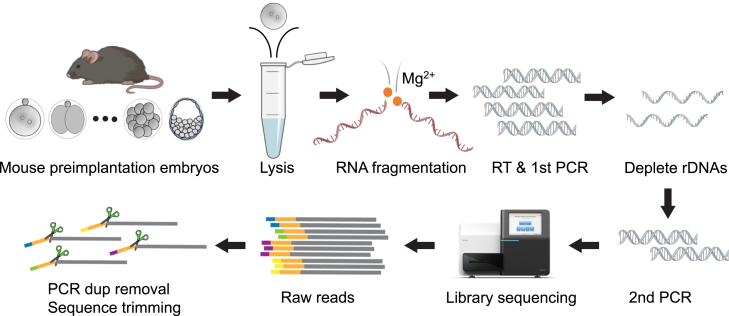
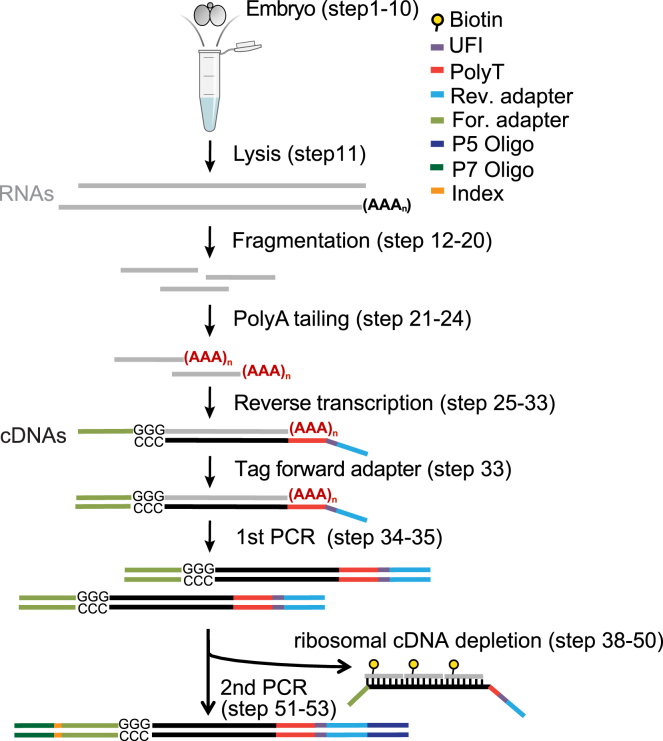
Strand-optimized Smart-seq (So-Smart-seq) can capture a comprehensive transcriptome from low-input samples. This technique detects both polyadenylated and non-polyadenylated RNAs, inclusive of repetitive RNAs, while excluding highly abundant ribosomal RNAs. So-Smart-seq preserves strand information and minimizes 5' to 3' coverage bias. We describe steps for the analysis of single mouse preimplantation embryos, including embryo isolation, library preparation, ribosomal cDNA depletion, and initial data processing. The protocol may be adapted for other low-input samples and the detection of small RNAs of <200 nt. For complete details on the use and execution of this protocol, please refer to Wei et al.1.
Keywords: Bioinformatics; Developmental biology; Genetics; Genomics; Molecular Biology; Systems biology.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.T.L. is a cofounder of Fulcrum Therapeutics, an advisor to Skyhawk Therapeutics, and a Non-Executive Director of the GSK.
Figures

References
-
- Behringer R. Cold Spring Harbor Laboratory Press; 2014. Manipulating the Mouse Embryo : A Laboratory Manual, Fourth Edition; pp. 137–148.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
