

Myotilin gene duplication causing late-onset myotilinopathy
- PMID: 39757377
- PMCID: PMC11702382
- DOI: 10.1111/ene.70029
Myotilin gene duplication causing late-onset myotilinopathy
Abstract
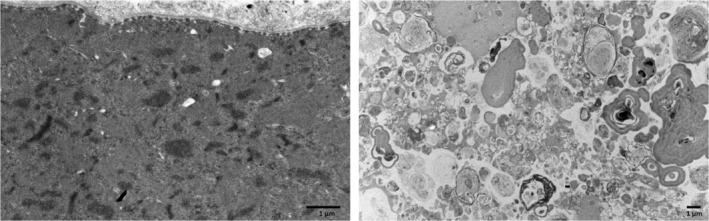
Background: myotilinopathy is a very rare inherited muscle disease that belongs to the group of myofibrillar myopathies. These diseases share a common alteration of the sarcomere organization at the level of the Z disk resulting in pathological protein aggregation, autophagic abnormalities, and ultimately muscle degeneration. Most reported cases are due to dominant missense mutations in the MYOT gene, two of which are largely recurrent.
Methods: We describe the clinical, radiological, pathological, and molecular analysis including long-read sequencing of a family affected by late-onset dominant proximodistal myopathy and muscle hypertrophy.
Results: We identified a duplication of the entire MYOT gene as the molecular cause of late-onset-myotilinopapthy with typical clinical and pathological features.
Conclusions: This study expands the molecular spectrum of myotilinopathy and highlights the use of long-read sequencing in the diagnosis of genetic neurological diseases caused by duplications and genomic structural variants. Myotilinopathy as well as other myofibrillar and distal myopathies should be considered in the differential diagnosis of patients affected by distal muscle weakness, even when presenting at an old age.
Keywords: distal myopathy; duplication; late‐onset myopathy; long‐read sequencing; muscle hypertrophy; myofibrillar myopathy; myotilin.
© 2025 The Author(s). European Journal of Neurology published by John Wiley & Sons Ltd on behalf of European Academy of Neurology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Nakano S, Engel A, Waclawik J, Emslie‐Smith A, Busis N. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol. 1996;55:549‐562. - PubMed
-
- Béhin A, Salort‐Campana E, Wahbi K, et al. Myofibrillar myopathies: state of the art, present and future challenges. Rev Neurol (Paris). 2015;171(10):715‐729. - PubMed
-
- Olivé M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain. 2005;128(10):2315‐2326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
