

Prospective identification of extracellular triacylglycerol hydrolase with conserved amino acids in Amycolatopsis tolypomycina's high G+C genomic dataset
- PMID: 39758972
- PMCID: PMC11697127
- DOI: 10.1016/j.btre.2024.e00869
Prospective identification of extracellular triacylglycerol hydrolase with conserved amino acids in Amycolatopsis tolypomycina's high G+C genomic dataset
Abstract
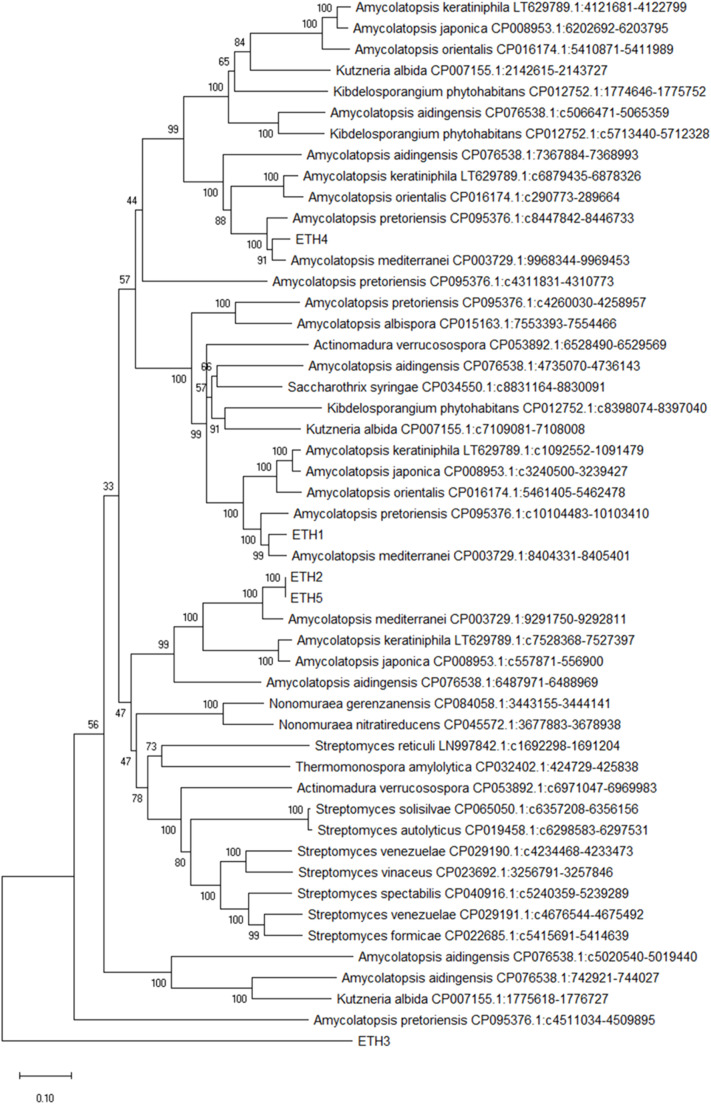
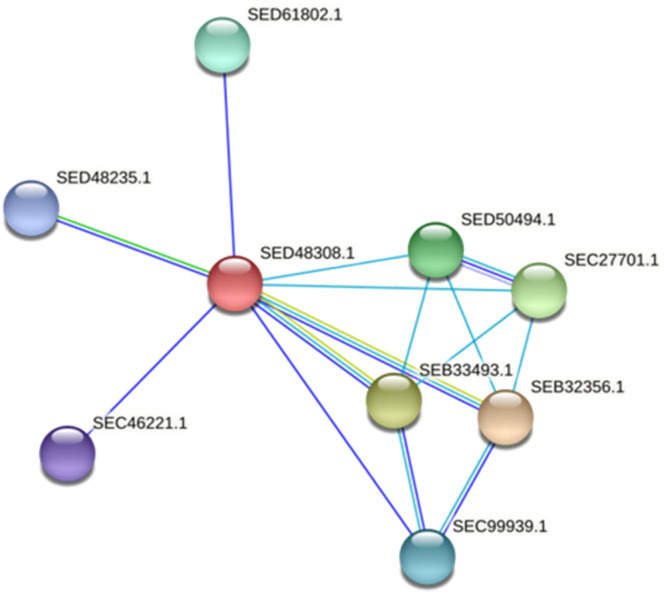
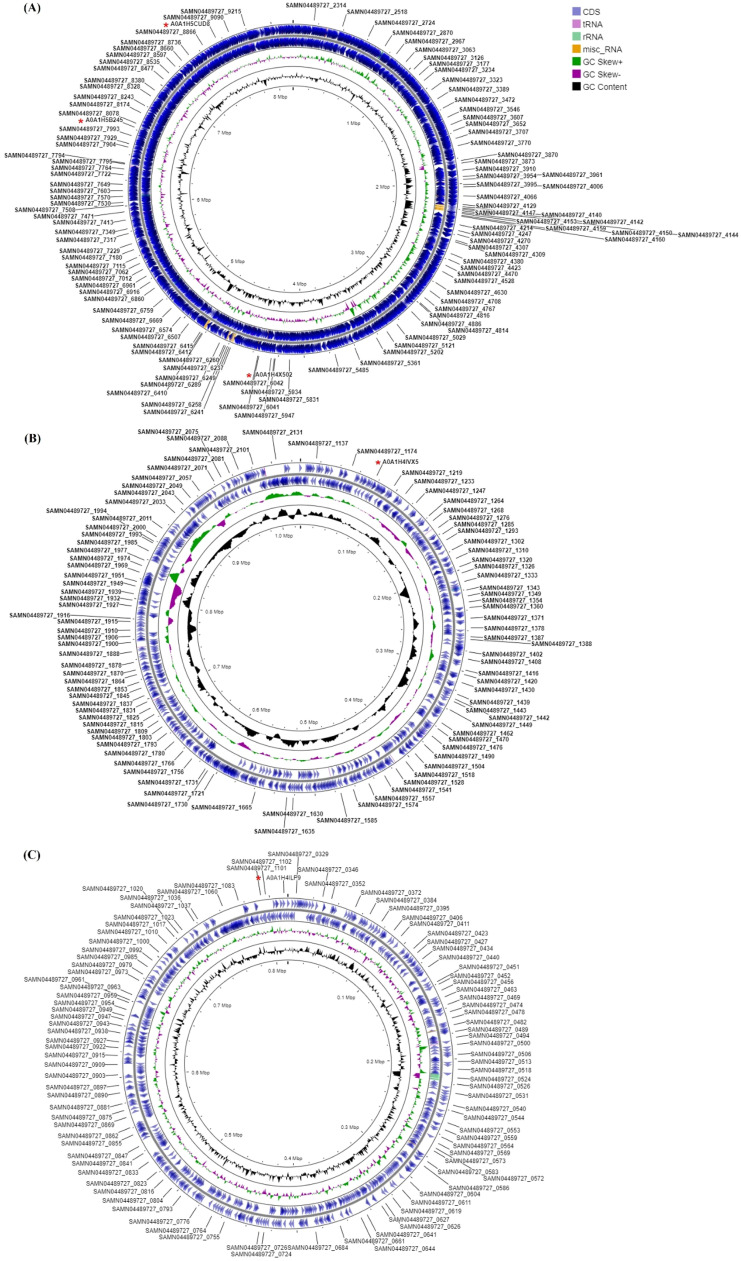
Extracellular triacylglycerol hydrolases (ETH) play a critical role for microorganisms, acting as essential tools for lipid breakdown and survival in challenging environments. The pursuit of more effective ETH genes and enzymes through evolution holds significant potential for enhancing living conditions. This study employs a proteogenomic approach to identify high G+C ETH in a notable Gram-positive bacterium, Amycolatopsis tolypomycina. Utilizing knowledge from genome and machine learning algorithms, prospective ETH genes/enzymes were identified. Notably, the ETH structural conserved accessibility to solvent clearly indicated the specific sixteen residues (GLY50, PRO93, GLY141, ASP148, GLY151, ASP172, ALA176, GLY195, TYR196, SER197, GLN198, GLY199, GLY200, GLY225, PRO327, ASP336) with no frequency. By pinpointing key residues and understanding their role, this study sets the stage for enhancing ETH performance through computational proteogenomic and contributes to the broader field of enzyme engineering, facilitating the development of more efficient and versatile ETH enzymes tailored to specific industrial or environmental contexts.
Keywords: Amycolatopsis tolypomycina; Computational proteogenomics; Conserved-solvent accessibility; Enzyme stability; Structural biology analysis.
© 2024 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures


References
-
- Abriata L.A., Dal Peraro M. State-of-the-art web services for de novo protein structure prediction. Brief. Bioinform. 2021;22:bbaa139. - PubMed
-
- Pucci F., Schwersensky M., Rooman M. Artificial intelligence challenges for predicting the impact of mutations on protein stability. Curr. Opin. Struct. Biol. 2022;72:161–168. - PubMed
-
- Veit-Acosta M., de A., Junior W.F. Computational prediction of binding affinity for cdk2-ligand complexes. A protein target for cancer drug discovery. Curr. Med. Chem. 2022;29:2438–2455. - PubMed
-
- Banaganapalli B., Anuradha C.M., Mulakayala C., D G., Chitta S. In silico structural characterization of mycobacterium tuberculosis h37rv udp-n-acetylmuramate dehydrogenase. Int. J. Integr. Biol. 2009;6:12–16.
LinkOut - more resources
Full Text Sources