

PRDM9 drives the location and rapid evolution of recombination hotspots in salmonid fish
- PMID: 39761307
- PMCID: PMC11703093
- DOI: 10.1371/journal.pbio.3002950
PRDM9 drives the location and rapid evolution of recombination hotspots in salmonid fish
Abstract
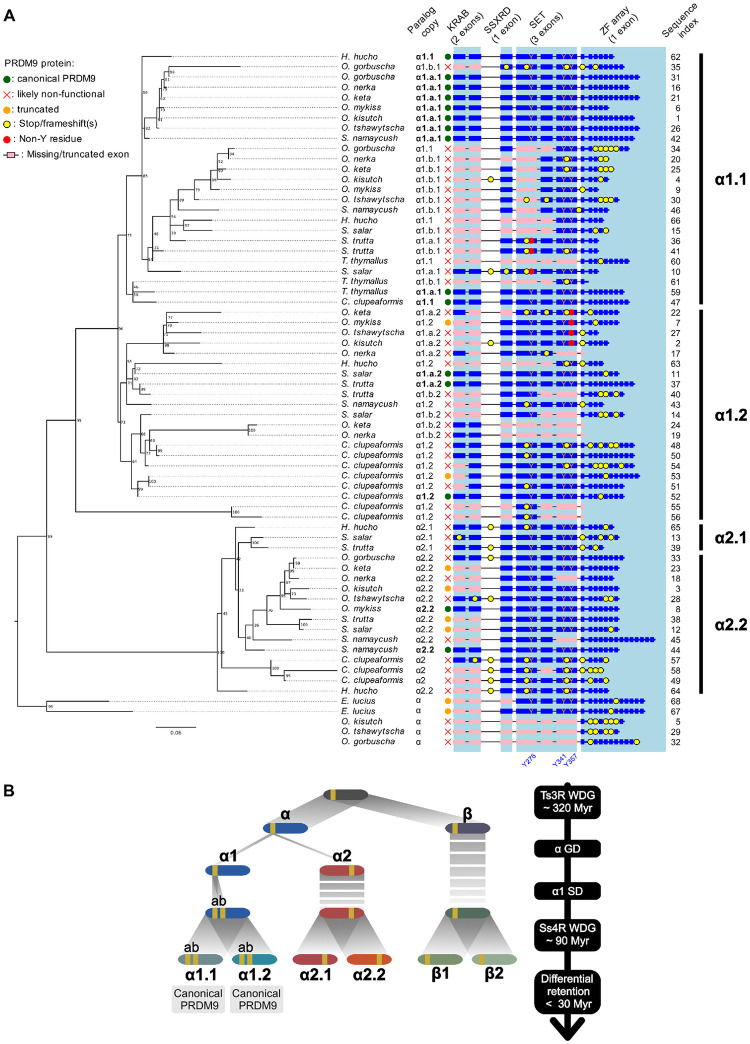
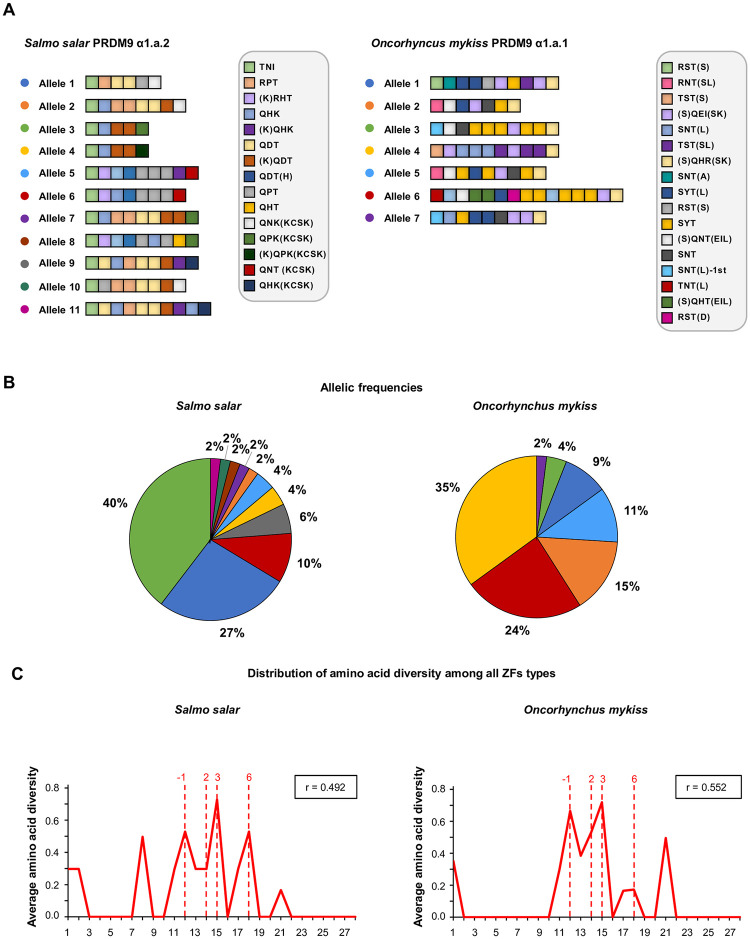
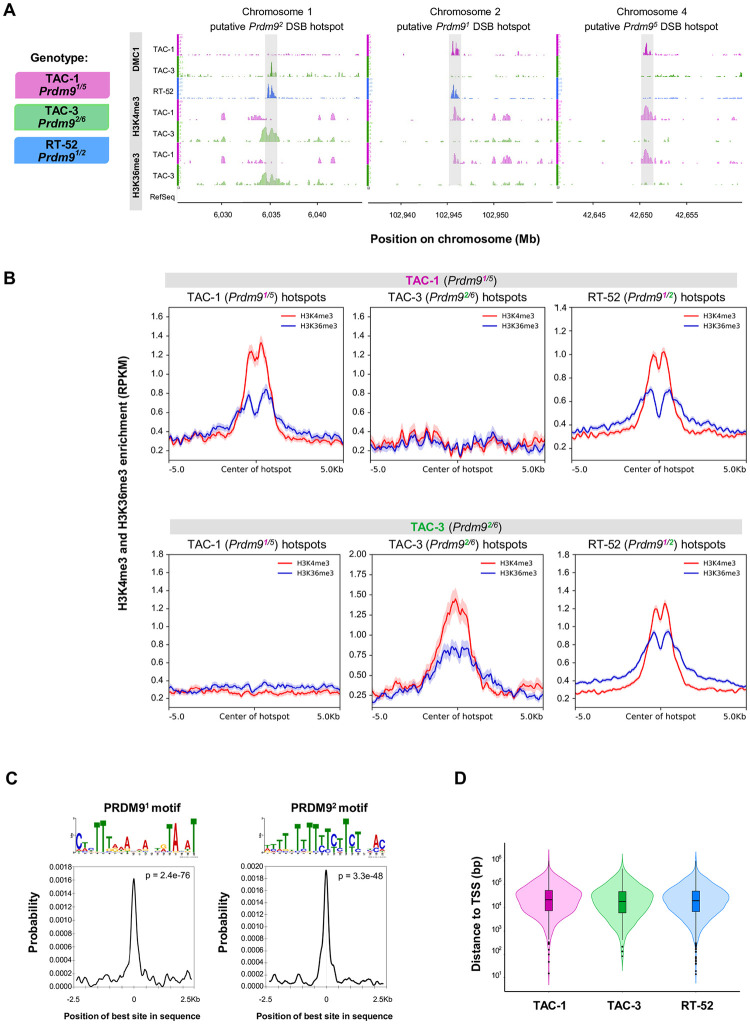
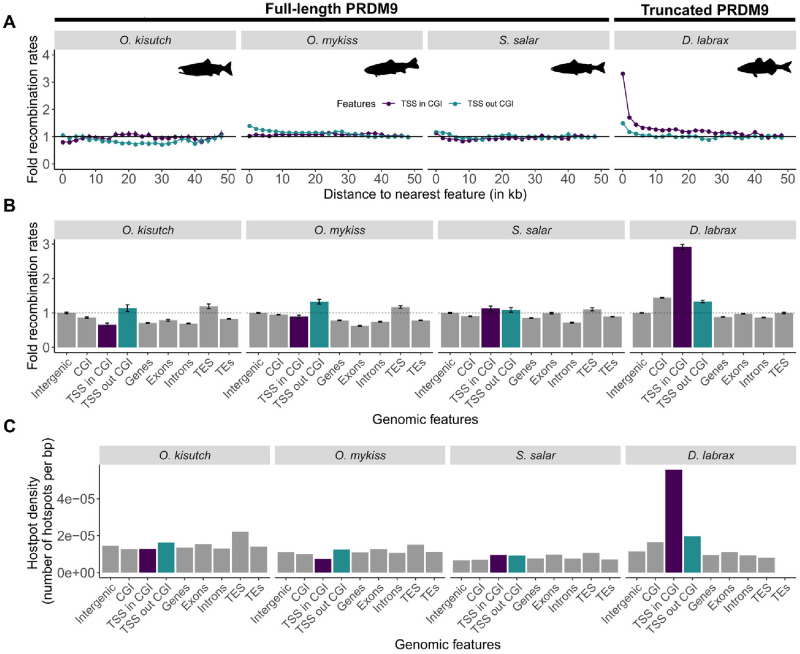
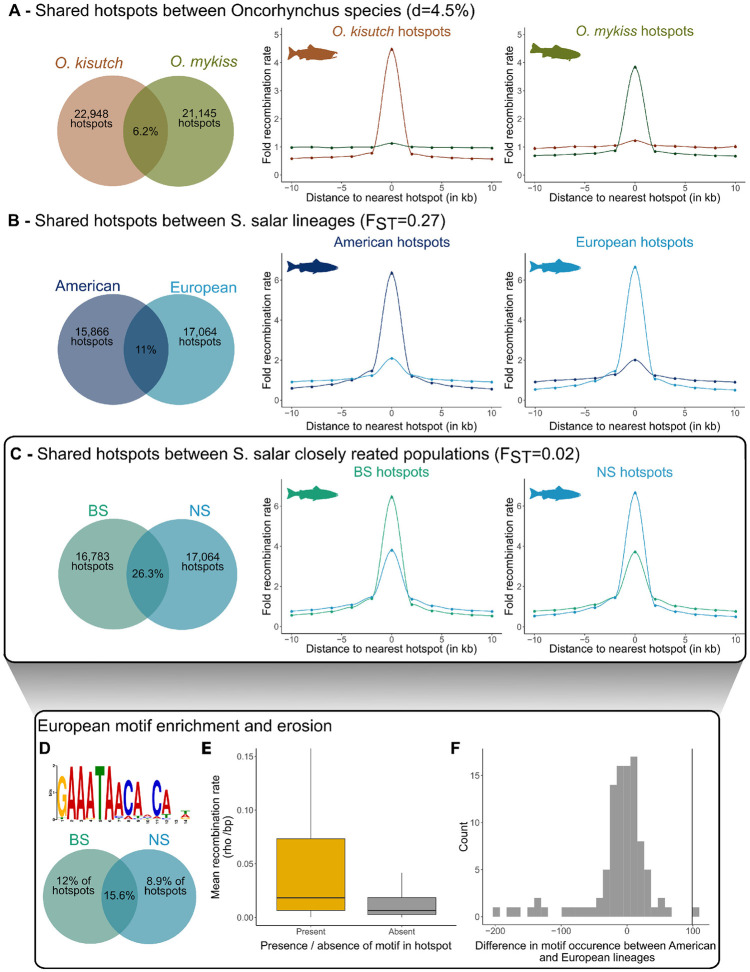
In many eukaryotes, meiotic recombination occurs preferentially at discrete sites, called recombination hotspots. In various lineages, recombination hotspots are located in regions with promoter-like features and are evolutionarily stable. Conversely, in some mammals, hotspots are driven by PRDM9 that targets recombination away from promoters. Paradoxically, PRDM9 induces the self-destruction of its targets and this triggers an ultra-fast evolution of mammalian hotspots. PRDM9 is ancestral to all animals, suggesting a critical importance for the meiotic program, but has been lost in many lineages with surprisingly little effect on meiosis success. However, it is unclear whether the function of PRDM9 described in mammals is shared by other species. To investigate this, we analyzed the recombination landscape of several salmonids, the genome of which harbors one full-length PRDM9 and several truncated paralogs. We identified recombination initiation sites in Oncorhynchus mykiss by mapping meiotic DNA double-strand breaks (DSBs). We found that DSBs clustered at hotspots positioned away from promoters, enriched for the H3K4me3 and H3K36me3 and the location of which depended on the genotype of full-length Prdm9. We observed a high level of polymorphism in the zinc finger domain of full-length Prdm9, indicating diversification driven by positive selection. Moreover, population-scaled recombination maps in O. mykiss, Oncorhynchus kisutch and Salmo salar revealed a rapid turnover of recombination hotspots caused by PRDM9 target motif erosion. Our results imply that PRDM9 function is conserved across vertebrates and that the peculiar evolutionary runaway caused by PRDM9 has been active for several hundred million years.
Copyright: © 2025 Raynaud et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Conserved features of recombination control in vertebrates.PLoS Biol. 2025 Jan 7;23(1):e3002959. doi: 10.1371/journal.pbio.3002959. eCollection 2025 Jan. PLoS Biol. 2025. PMID: 39775336 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
