

Spatial tumor immune heterogeneity facilitates subtype co-existence and therapy response in pancreatic cancer
- PMID: 39762215
- PMCID: PMC11704331
- DOI: 10.1038/s41467-024-55330-7
Spatial tumor immune heterogeneity facilitates subtype co-existence and therapy response in pancreatic cancer
Abstract
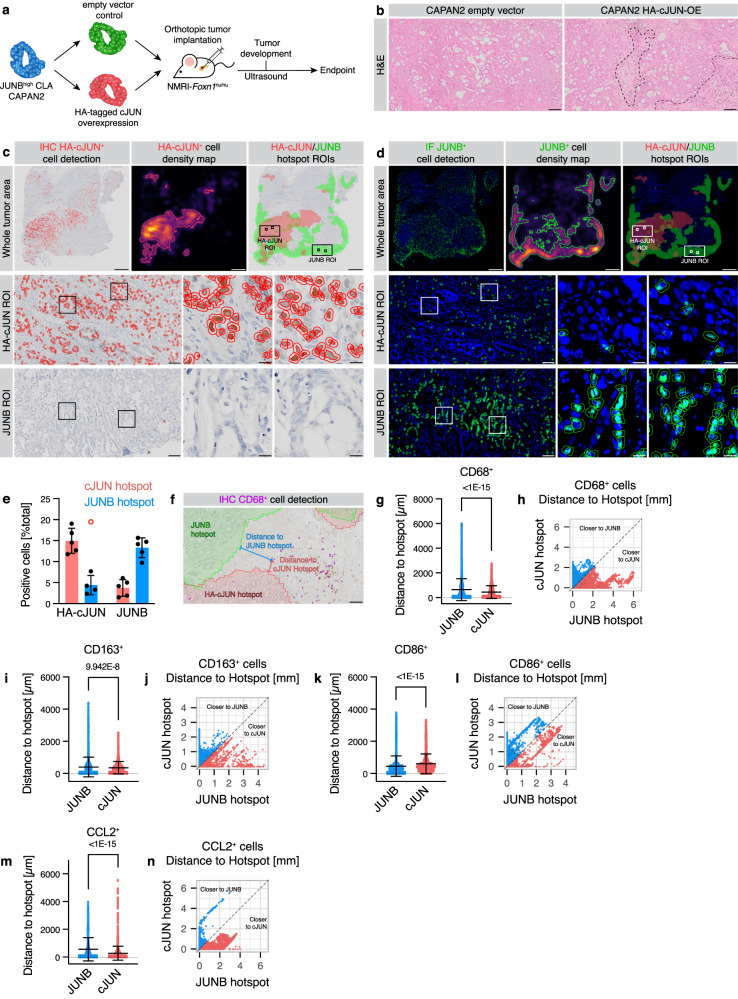
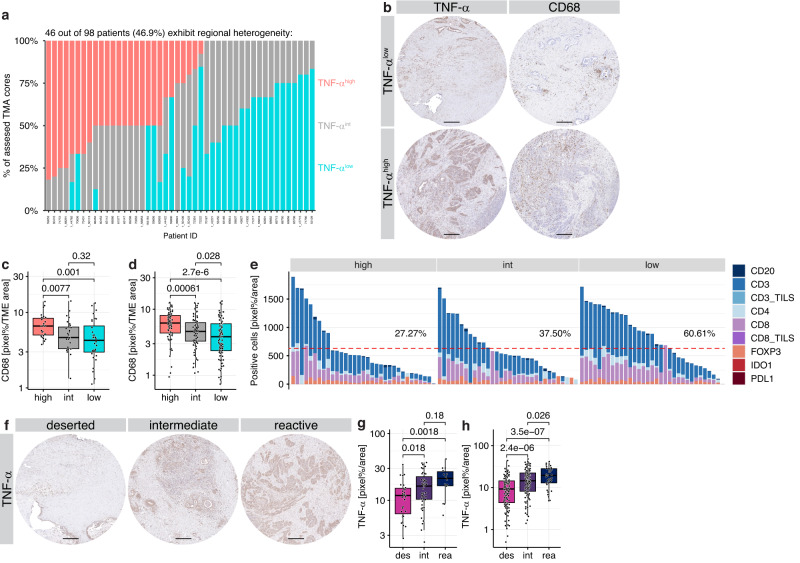
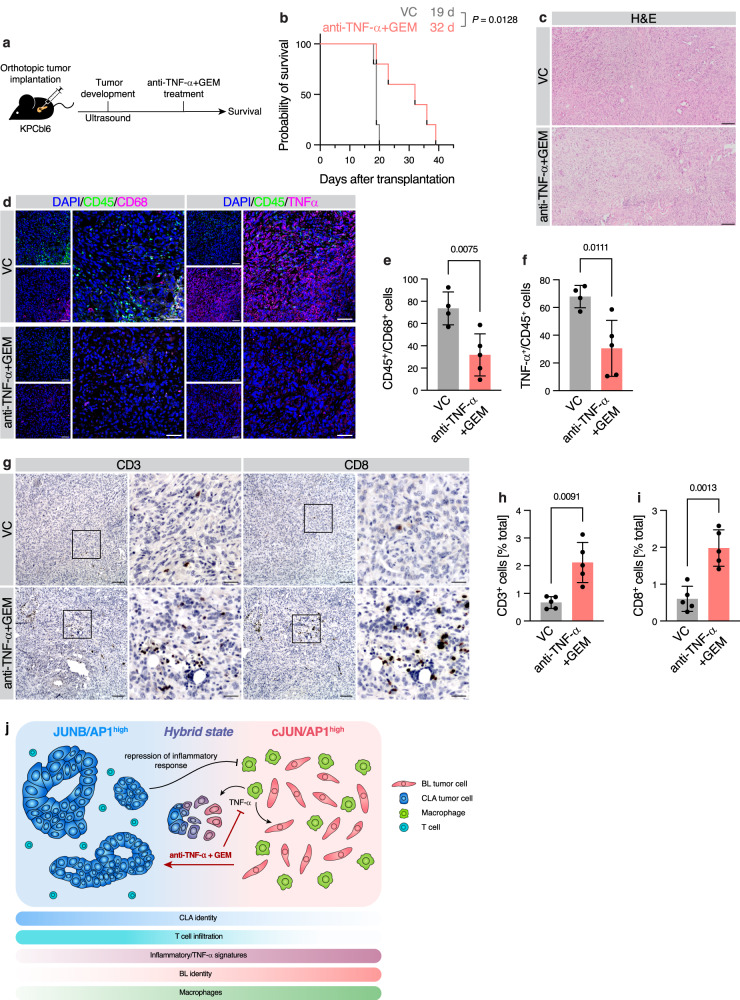
Pancreatic ductal adenocarcinoma (PDAC) displays a high degree of spatial subtype heterogeneity and co-existence, linked to a diverse microenvironment and worse clinical outcome. However, the underlying mechanisms remain unclear. Here, by combining preclinical models, multi-center clinical, transcriptomic, proteomic, and patient bioimaging data, we identify an interplay between neoplastic intrinsic AP1 transcription factor dichotomy and extrinsic macrophages driving subtype co-existence and an immunosuppressive microenvironment. ATAC-, ChIP-, and RNA-seq analyses reveal that JUNB/AP1- and HDAC-mediated epigenetic programs repress pro-inflammatory signatures in tumor cells, antagonizing cJUN/AP1 signaling, favoring a therapy-responsive classical neoplastic state. This dichotomous regulation is amplified via regional TNF-α+ macrophages, which associates with a reactive phenotype and reduced CD8+ T cell infiltration in patients. Consequently, combined preclinical anti-TNF-α immunotherapy and chemotherapy reduces macrophages and promotes CD3+/CD8+ T cell infiltration in basal-like PDAC, improving survival. Hence, tumor cell-intrinsic epigenetic programs, together with extrinsic microenvironmental cues, facilitate intratumoral subtype heterogeneity and disease progression.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin.73, 17–48 (2023). - PubMed
-
- Sohal, D. P. S. et al. Metastatic pancreatic cancer: ASCO guideline update. JCO38, 3217–3230 (2020). - PubMed
-
- Surveillance Research Program, National Cancer Institute. SEER*Explorer: An interactive website for SEER cancer statistics. Data source(s): SEER Incidence Data, November 2022 Submission (1975-2020).
-
- Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature531, 47–52 (2016). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
- 70112999/Deutsche Krebshilfe (German Cancer Aid)
- 70115054/Deutsche Krebshilfe (German Cancer Aid)
- KFO5002/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 10.20.2.038MN; 10.23.2.021MN/Fritz Thyssen Stiftung (Fritz Thyssen Foundation)
- 2021.159.1 and 2021.159.2/Wilhelm Sander-Stiftung (Wilhelm Sander Foundation)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
