

NLRP3 inflammasome-modulated angiogenic function of EPC via PI3K/ Akt/mTOR pathway in diabetic myocardial infarction
- PMID: 39762890
- PMCID: PMC11705910
- DOI: 10.1186/s12933-024-02541-3
NLRP3 inflammasome-modulated angiogenic function of EPC via PI3K/ Akt/mTOR pathway in diabetic myocardial infarction
Abstract
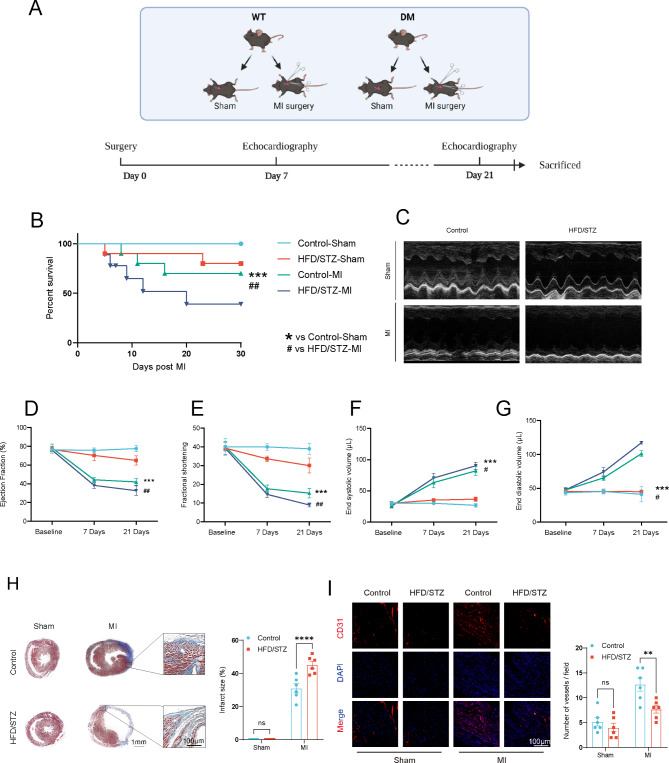
Background: Inflammatory diseases impair the reparative properties of endothelial progenitor cells (EPC); however, the involvement of diabetes in EPC dysfunction associated with myocardial infarction (MI) remains unknown.
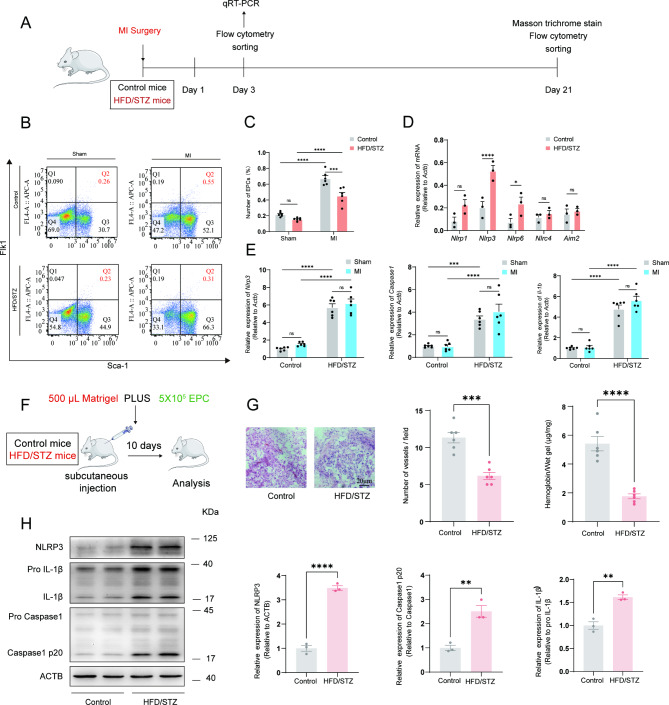
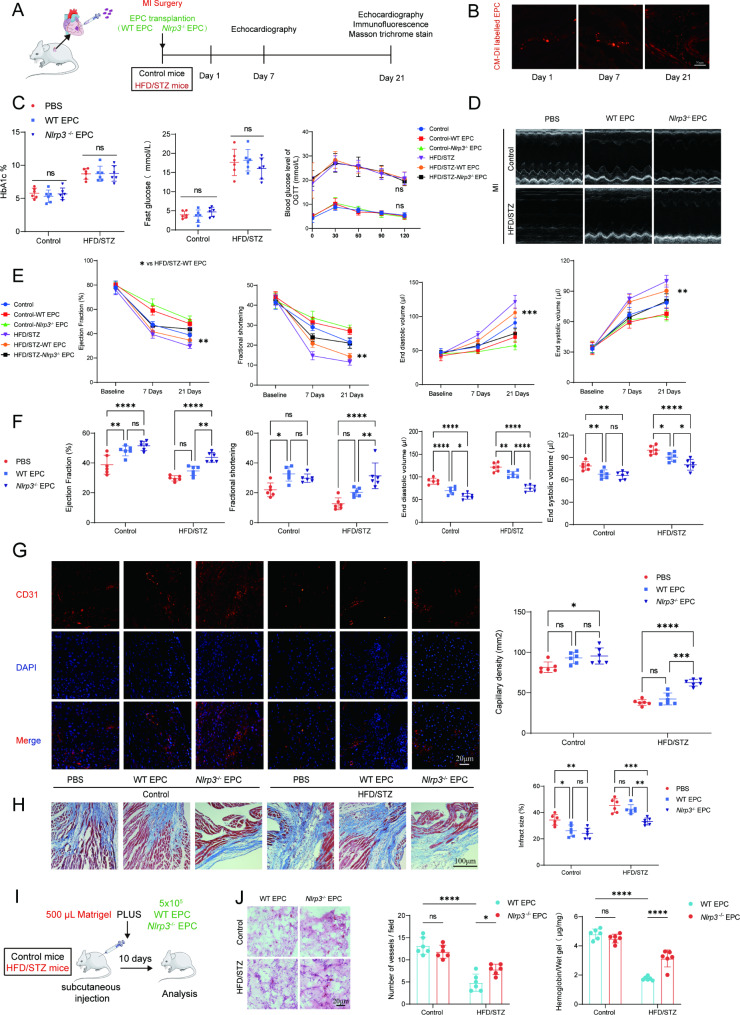
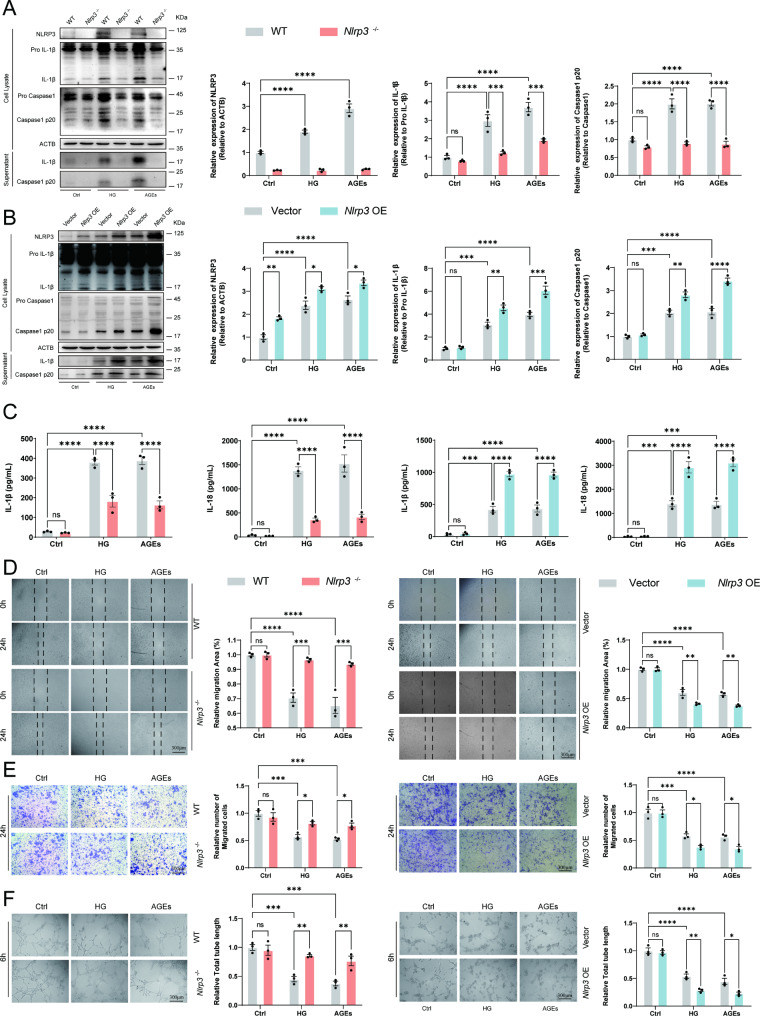
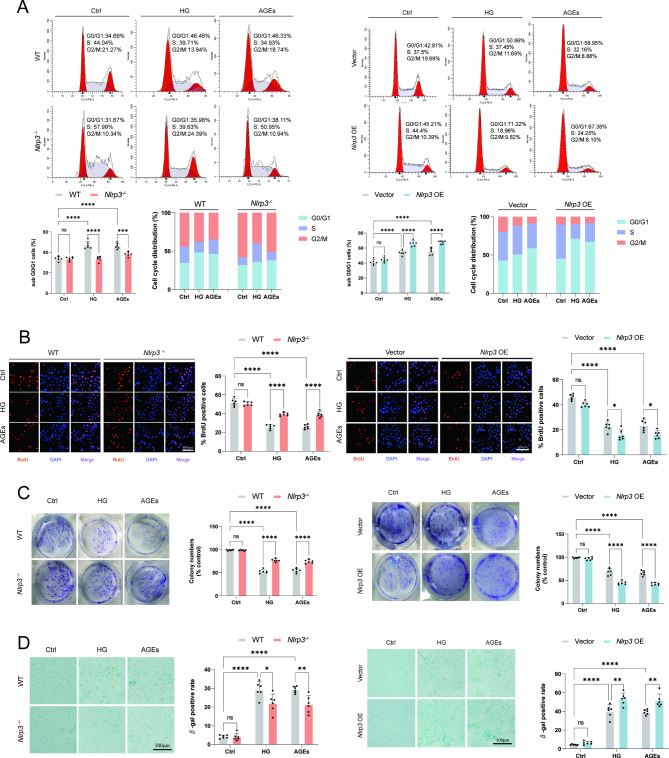
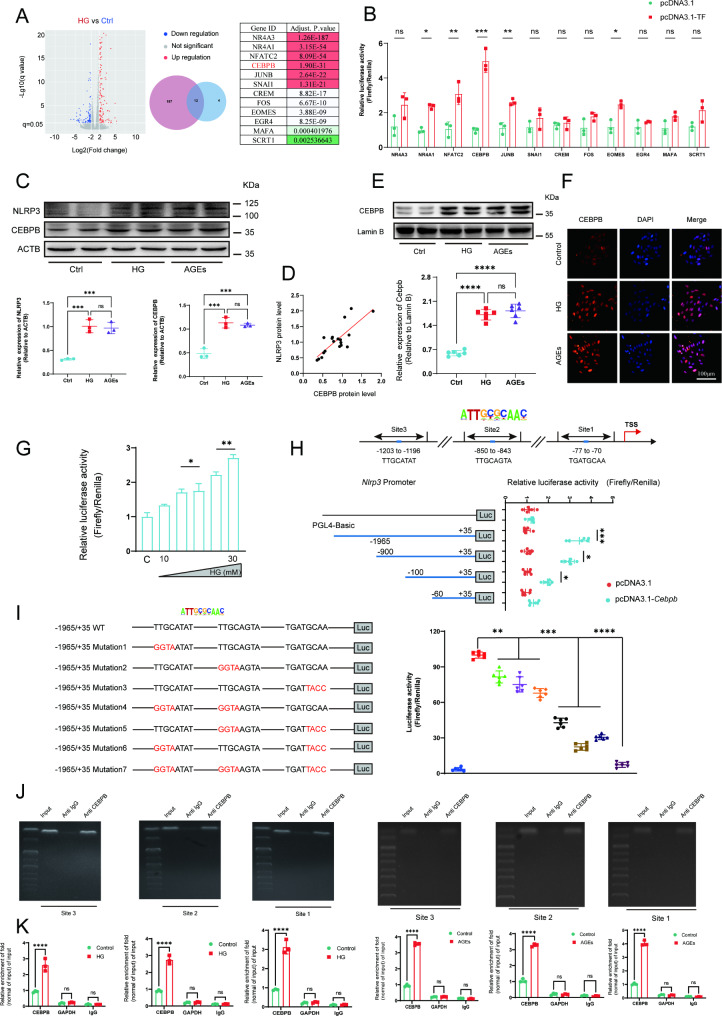
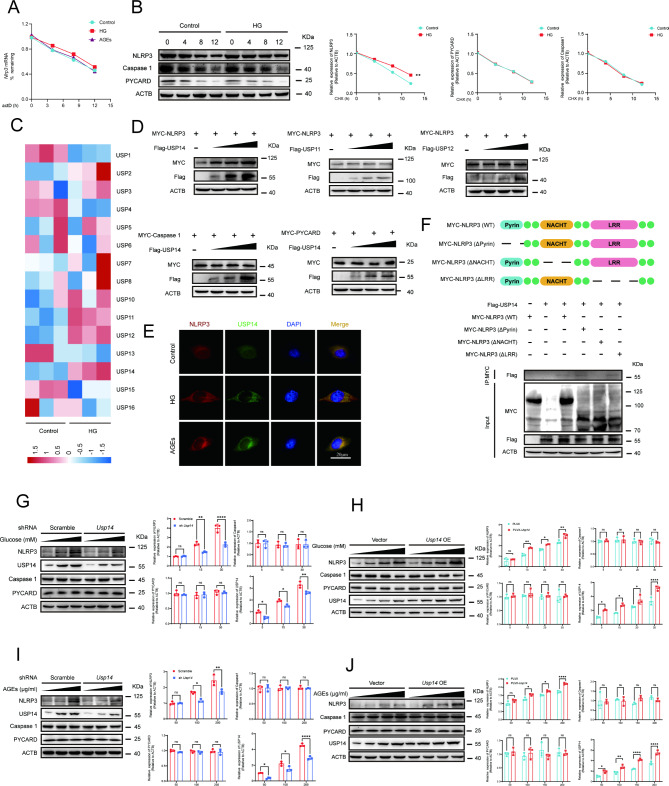
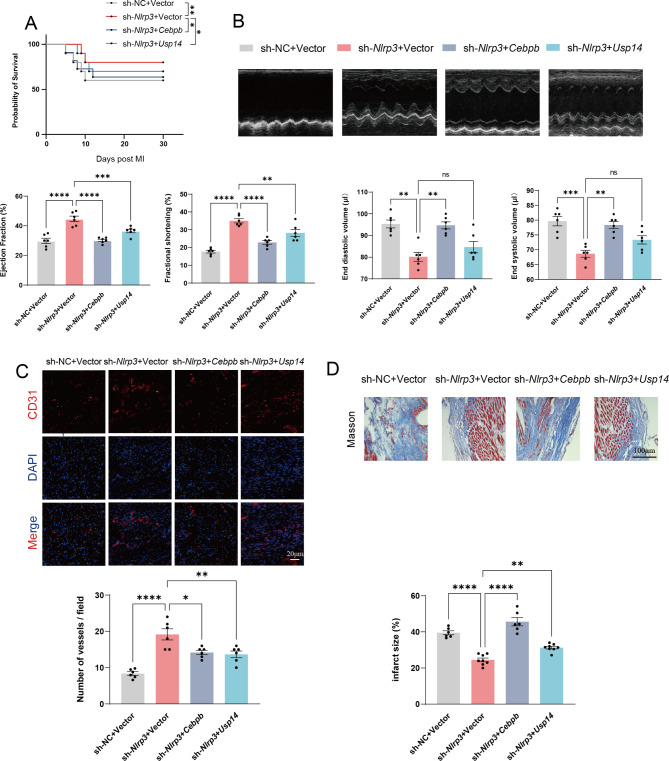
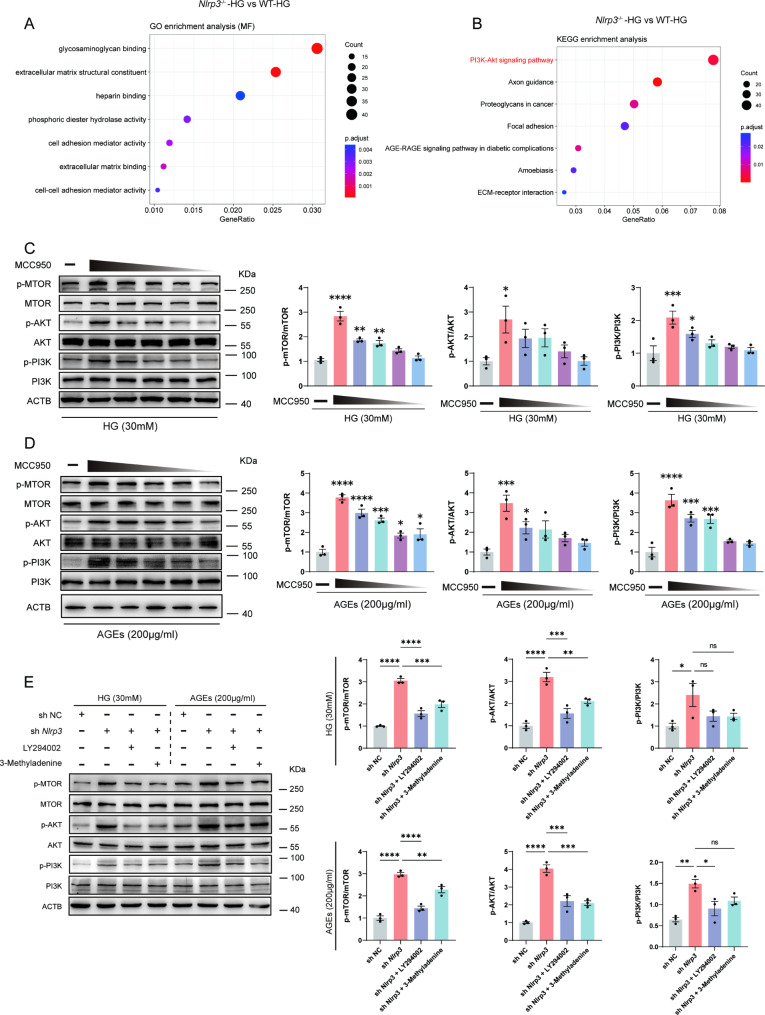
Methods: A model was established combining high-fat diet (HFD)/streptozotocin (STZ)-induced diabetic mice with myocardial infarction. The therapeutic effects of transplanted wild-type EPC, Nlrp3 knockout EPC, and Nlrp3 overexpression EPC were evaluated. Chip and Luciferase assay revealed CEBPB regulated the transcriptional expression of Nlrp3 as a transcription factor in EPC stimulated by high glucose (HG) or advanced glycation end products (AGEs). CO-IP results suggested that USP14 selectively suppressed NLRP3 degradation. KEGG enrichment revealed PI3K/ Akt/mTOR signaling showed striking significance in the entire pathway.
Results: In our study, wild-type, Nlrp3 knockout and Nlrp3 overexpressed EPC, intracardiac injections effectively improved cardiac function, increased angiogenesis, and reduced infarct size in mice with myocardial infarction. However, in the HFD/STZ-induced diabetic mice model combined with myocardial infarction, Nlrp3 knockout EPC significantly restored angiogenic capacity. Mechanically, CEBPB regulated the transcriptional level of Nlrp3 as a transcription factor in EPC. Meanwhile, we found that USP14 selectively suppressed NLRP3 protein degradation through the USP motif on the NACHT domain in mediating inflammasome activation. Cardiac functional outcomes in recipient mice after intramyocardial injection of shNlrp3 EPC overexpressing CEBPB or USP14 validated the modulation of EPC function by regulating Nlrp3 transcription or post-translational modification. Furthermore, KEGG enrichment and validation at the protein levels revealed PI3K/ Akt/mTOR cascade might be a downstream signal for NLRP3 inflammasome.
Conclusion: Our study provides a new understanding of how diabetes affected progenitor cell-mediated cardiac repair and identifies NLRP3 as a new therapeutic target for improving myocardial infarction repair in inflammatory diseases.
Keywords: Angiogenesis; Diabetes; Endothelial progenitor cell; Myocardial infarction; NLRP3 inflammasome.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Consent for publication: All authors consent for the publication of this study. Competing interests: The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
