

This is a preprint.
A precision oncology-focused deep learning framework for personalized selection of cancer therapy
- PMID: 39763776
- PMCID: PMC11702554
- DOI: 10.1101/2024.12.12.628190
A precision oncology-focused deep learning framework for personalized selection of cancer therapy
Abstract
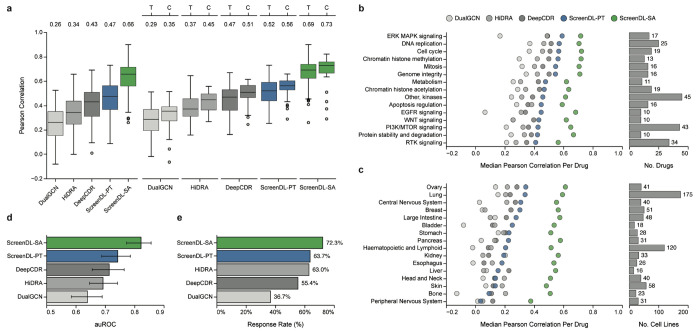
Precision oncology matches tumors to targeted therapies based on the presence of actionable molecular alterations. However, most tumors lack actionable alterations, restricting treatment options to cytotoxic chemotherapies for which few data-driven prioritization strategies currently exist. Here, we report an integrated computational/experimental treatment selection approach applicable for both chemotherapies and targeted agents irrespective of actionable alterations. We generated functional drug response data on a large collection of patient-derived tumor models and used it to train ScreenDL, a novel deep learning-based cancer drug response prediction model. ScreenDL leverages the combination of tumor omic and functional drug screening data to predict the most efficacious treatments. We show that ScreenDL accurately predicts response to drugs with diverse mechanisms, outperforming existing methods and approved biomarkers. In our preclinical study, this approach achieved superior clinical benefit and objective response rates in breast cancer patient-derived xenografts, suggesting that testing ScreenDL in clinical trials may be warranted.
Conflict of interest statement
Competing Interests Statement University of Utah may license the models described herein to for-profit companies, which may result in tangible property royalties to members of the Welm labs who developed the models. M.T.L. is a Manager in StemMed Holdings L.L.C., a limited partner in StemMed Ltd., and holds an equity stake in Tvardi Therapeutics. The other authors declare no conflicts.
Figures












References
-
- Haslam A., Kim M. S. & Prasad V. Updated estimates of eligibility for and response to genome-targeted oncology drugs among US cancer patients, 2006-2020. Ann. Oncol. 32, 926–932 (2021). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources